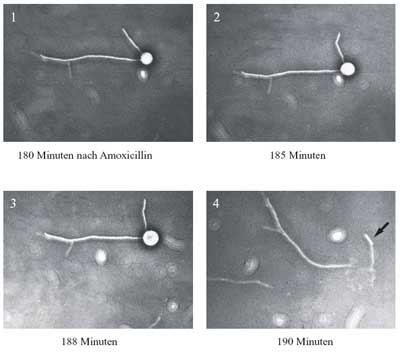
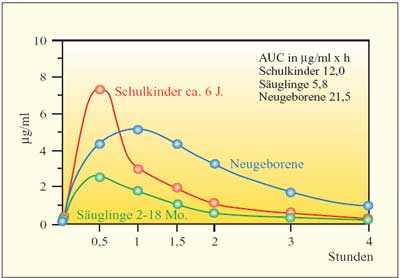
| Die
einzelnen Präparate Penicilline
Nach
ihrer chemischen Struktur sind alle Penicilline Derivate der
6-Aminopenicillansäure. Die Penicilline sind als schwache
Säuren unbeständig, stabiler sind die neutralen Salze
und die Ester, welche auch gut wasserlöslich sind. An die
Seitenkette werden Radikale angehängt, die über die
antimikrobielle Wirksamkeit entscheiden.
Benzylpenicillin
(Penicillin G) ist nach wie vor das wirksamste Antibiotikum
gegen ß-hämolysierende Streptokokken. Mit einem MHK-Wert
von 0,01–0,001 µg/ml ist die Empfindlichkeit dieses
Erregers auch nach > 60 Jahren unübertroffen.
Das
Benzylpenicillin ist als leichtwasserlösliches Natrium-
oder Kaliumsalz oder als schwer lösliches Depotpenicillin
(Procain-Penicillin G, Clemizol-Penicillin G oder Benzathin-Penicillin
G) verfügbar. Es liegt als Benzylpenicillin-Natrium zur
parenteralen Verabreichung vor. Die Tagesdosis beträgt
je nach Schwere der Infektion im Kindesalter zwischen 100 000
iE/kg KG bis 400 000 iE/kg KG bei der eitrigen Meningitis. Da
die Halbwertszeit des intravenös verabreichten Penicillin
G nur ca. 30 Minuten beträgt, sind 6-stündliche Dosierungsintervalle
(Aufteilung der Tagesdosis auf mindestens 4 Dosen) nötig.
Auch intramuskulär verabreichtes Benzylpenicillin hat eine
ähnliche kurze Halbwertszeit und eignet sich, da es ebenso
6-stündlich intramuskulär verabreicht werden müsste,
nur in Kombination mit Depotpenicillinen, um einen raschen Wirkspiegel
zu erreichen.
Procain-
oder Clemizolpenicillin werden intramuskulär verabreicht
und haben einerseits einen langsamen Konzentrationsanstieg mit
niedrigen Konzentrationen zur Folge, der mit einer Halbwertszeit
von 6–8 Stunden abfällt.
Nach
intramuskulärer Gabe von Benzathin-Penicillin G erreicht
man maximale Serumkonzentrationen von 0,1 µg/ml, jedoch
über einen Zeitraum von 20–25 Tagen. Dadurch eignet
sich dieses Präparat primär zur Dauerprophylaxe bei
rheumatischem Fieber 1 x monatlich. Auch zur Behandlung der
exquisit empfindlichen Spirochaeten kann Benzathin-Penicillin
eingesetzt werden.
Klinische
Indikation für intra-venöses Penicillin G-Natrium
Die parenterale Verbreichung von Penicillin G ist bei schweren
bakteriellen Infektionen mit Mikroorganismen mit bekannter Empfindlichkeit,
wie Pneumokokken, Meningokokken, Streptokokken, Leptospiren,
Aktinomyzes, Gasbrand, Tetanus, indiziert. Die Behandlung erfordert
wegen des schweren Krankheitsbildes eine stationäre Aufnahme,
wobei Verabreichungsintervalle von 6 Stunden wegen der kurzen
Halbwertszeit eingehalten werden können. Entsprechende
Krankheitsbilder sind ein Waterhouse-Friderichsen-Syndrom, ein
toxischer Scharlach, ein Erysipel, ein rheumatisches Fieber
initial bis zur klinischen Stabilisierung, Gasbrand und Tetanus.
Bei diesen Krankheitsbildern ist durch eine Prima-Vista-Diagnose
der Verdacht auf einen Penicillin-empfindlichen Keim gegeben
(Abbildung 7a, 7b).
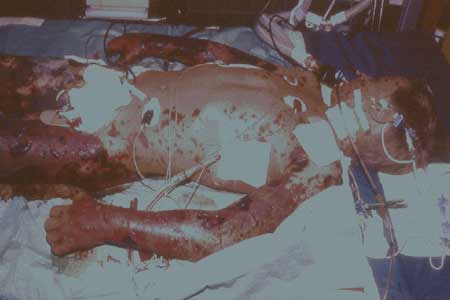
Abbildung
7a: Waterhouse-Friderichsen-Syndrom
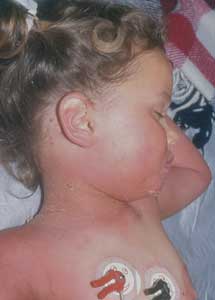
Abbildung
7b: toxischer Scharlach
Die
bakterielle Endocarditis kann mit Penicillin in Kombination
mit einem Aminoglykosid behandelt werden, bis die Erregerdiagnose
mit den entsprechenden Empfindlichkeiten vorliegt.
Bei
einer akut beginnenden Lobärpneumonie kann man mit einem
parenteralen Penicillin G beginnen. Eine Pneumonie kann jedoch
durch verschiedene Mikroorganismen verursacht werden, die nicht
alle auf Penicillin empfindlich sind. Die Erregerdiagnose mit
Resistenzprüfung dauert meist Tage. Schnelltests wie der
Nachweis von Pneumokokkenantigenen im Harn sind zwar möglich,
der Test ist aber nicht breit verfügbar und sagt nichts
über die Empfindlichkeit der Pneumokokken aus.
Aus
diesem Grund ist die Verabreichung eines Breitspektrum-Penicillins
oder Cephalosporins der Cefuroxim- oder der Cefotaximgruppe
bei einer ambulant erworbenen Pneumonie vorzuziehen (Abbildung
8a). Bei Verdacht auf eine atypische Pneumonie ist die Behandlung
mit einem Makrolid-Antibiotikum zu beginnen. Bei vollkommener
Unklarheit ist im Kindesalter eine Kombination eines Breitspektrum-Cephalosporins
mit einem Makrolid angezeigt, bei Jugendlichen wird ein Chinolon
verabreicht. Dies gilt nicht für die Streptokokken B-Pneumonie
± Sepsis im Neugeborenenalter, die auch weitgehend klinisch
festzumachen ist und mit Penicillin G – evtl. in Kombination
mit einem Aminoglykosid – behandelt wird (Abbildung 8b).
Zur Prophylaxe einer Streptokokken B-Infektion beim Neugeborenen
kann einer Streptokokken-positiven Mutter 2 Stunden präpartal
200 000 iE eines Depotpenicillins i.m. verabreicht werden.
Abbildung
8a: Typische Lobärpneumonie durch Pneumokokken
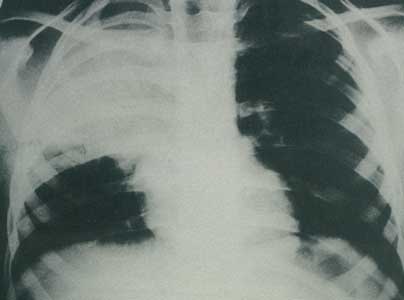
Abbildung
8b: Streptokokken B-Pneumonie und Sepsis bei einem
reifen Neugeborenen
Phenoxymethylpenicillin
Die Besonderheit von Phenoxymethylpenicillin V ist die Widerstandsfähigkeit
gegen Zerstörung durch die Magensäure. Säurestabil
sind neben dem Phenoxymethylpenicillin (Penicillin V) auch Propicillin
und Azidocillin, die jedoch in der Kinderärztlichen Praxis
eine untergeordnete Rolle spielen, nicht zuletzt da Propicillin
eine um das 5fache schwächere Wirksamkeit gegen Gram-positive
Penicillin-empfindliche Erreger besitzt und sowohl Propicillin
als auch Azidocillin mit 80 – 85% Eiweißbindung
deutlich niedrigere freie Wirkstoffkonzentrationen im Serum
und im Gewebe erreichen als nach Gabe von Phenoxymethylpenicillin
V, das eine Eiweißbindung von 20% aufweist. Die orale
Gabe von Penicillin G ist wegen der fehlenden Säurestabilität
nicht sinnvoll.
Die
Tagesdosis beträgt bei Kindern 100 000 iE/kg KG, die man
auch beim Benzathinsalz am ersten Tage in 8-stündigen Intervallen
verabreichen sollte, danach wird die Tagesdosis auf 2 Dosen
verteilt. Bei Durchfall zu Beginn der Behandlung ist eine einmalige
Dosis eines Depotpenicillins intramuskulär günstig,
da eine ausreichende Resorption eines Oralpenicillins nicht
gewährleistet ist. Anschließend, d.h. spätestens
nach 36 Stunden, muss mit einem Oralpenicillin fortgesetzt werden,
um einen 10-Tages-Wirkspiegel aufrechtzuerhalten.
Der
klinische Einsatz eines Oralpenicillins beruht in erster Linie
auf der Behandlung der Streptokokkenangina. Die Diagnose beruht
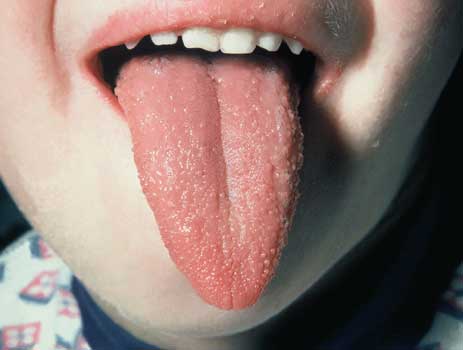
auf klinischen Kriterien wie einem scarlatiniformen Exanthem
und der Himbeerzunge (Abbildung 9), die erst nach 24 Stunden
erscheint. Differenzialdiagnostische Hinweise gibt die Anamnese
und die epidemiologische Situation. Klinisch präsentiert
sich eine Streptokokkenangina mit einem schweren Krankheitsbild
mit plötzlichem Beginn, mit hohem Fieber und Schluckschmerzen.
Typisch sind Brechreiz und Erbrechen, Bauchschmerzen und geschwollene,
druckempfindliche Halslymphknoten. Der Tonsillenbefund zeigt
initial hochrote glasig geschwollene Tonsillen ohne Beläge.
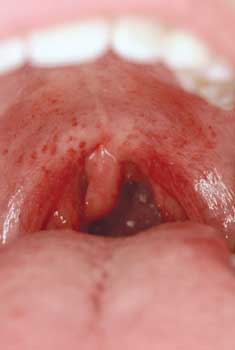
Pathognomonisch sind Petechien am weichen Gaumen (Abbildung
10). Wenn eine Streptokokkenangina nicht antibiotisch behandelt
wird, treten ab dem 2.–3.Tag flache Ulzerartionen auf
den Tonsillen auf.
Abbildung
9: Himbeerzunge bei Scharlach
Abbildung
10: Petechien am weichen Gaumen typisch bei Scharlach
Die
Diagnose kann durch den Streptokokken-Schnelltest bestätigt
werden.
Zahlreiche
Studien zeigen, dass nur eine Therapiedauer von 10 Tagen zu
einer klinischen und „bakteriologischen Heilung“
mit Eradikation von Streptokokken in >95% der Patienten führt.
Bemerkenswert und bis vor kurzem unklar war, dass Streptokokken,
die trotz einer 10-tägigen Penicillinbehandlung noch in
3–8% der Patienten im Rachen isoliert werden können,
weiterhin voll Penicillin-empfindlich sind. Untersuchungen zeigten,
dass Streptokokken auch von sog. „nicht professionellen
Phagozyten“ wie Epithelzellen aufgenommen werden können
und intrazellulär überleben, ohne die Wirtszelle zu
zerstören. Nach 10, 15, 18 Tagen werden auch diese Streptokokken
aus den Epithelzellen freigesetzt und werden im Rachenabstrich
nachgewiesen. Die so überlebenden Streptokokken haben jedoch
ihre Virulenz, d.h. Infektiosität, verloren und sind als
apathogen zu betrachten.
Oralpenicilline
können auch zur frühzeitigen Behandlung einer Borrelieninfektion,
eines Erythema chronicum migrans eingesetzt werden. Die Behandlungsdauer
beträgt 14 Tage, es sind streng 8-stündliche Dosisintervalle
einzuhalten, um einen kontinuierlichen Wirkstoffspiegel aufrechtzuerhalten
(Abbildung 11).
Abbildung
11: Erythema chronicum migrans nach Zeckenstich
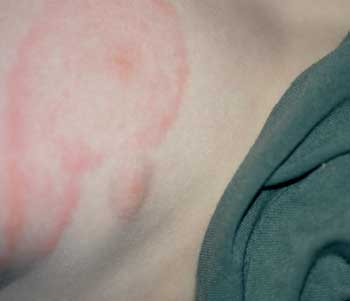
Auch
bei peridontalen Infektionen sind Oralpenicilline indiziert,
wie auch zu einer Endocarditis-Prophylaxe bei Herzklappenfehlern,
bei Zahnbehandlung, bzw. bei der Prophylaxe des rheumatischen
Fiebers.
Die
Impetigo contagiosa ist eine Mischinfektion von Streptococcus
pyogenes und meist ß-Lactamase-bildenden Staphylokokken.
Dabei kommt aber der pathologischen transienten Streptokokkenflora
die größere pathogenetische Bedeutung zu. Diese kann
meist mit einem Oralpenicillin erfolgreich behandelt werden.
Die Behandlung einer Impetigo contagiosa erfolgt auch um eine
hämorrhagische Poststreptokokken-Glomerulonephritis zu
verhindern, die bei besonderen nephritogenen Streptokokkenstämmen
auftreten kann.
ß-Lactamase-stabile
Penicillinabkömmlinge, Isoxazolylpenicilline
Isoxazolylpenicilline sind gegen ß-Lactamasen von Staphylokokken
stabil. Sie haben prinzipiell den gleichen Wirkmechanismus wie
Penicillin und sind weitgehend gegen die gleichen Gram-positiven
Mikroorganismen wirksam, allerdings um den Faktor 10 geringer
als Penicillin. Isoxazolylpenicilline werden meist parenteral
verabreicht, die orale Verabreichung ist möglich. Die enterale
Resorption ist am besten bei Flucloxacillin, dieses Präparat
ist auch in Saftform erhältlich. Alle Isoxazolylpenicilline
werden nach Nahrung deutlich schlechter resorbiert. Kritisch
ist auch die hohe Serum-Eiweißbindung zu bewerten, die
zwischen 95% und 98% liegt.
Die
Präparate sind bei Penicillin-Allergie kontraindiziert,
die übrigen unerwünschten Wirkungen entsprechen weitgehend
denen der anderen Penicilline. Cholestatische Hepatitiden kommen
häufiger nach Gabe von Flucloxacillin als nach Oxacillin
oder Dicloxacillin vor. Bei eingeschränkter Nierenfunktion
wird je nach Kreatinin-Clearance das Dosierungsintervall auf
8 bzw. 12 Stunden verlängert.
Die
Präparate Oxacillin, Cloxacillin und Flucloxacillin sind
ausschließlich bei schweren Infektionen durch Penicillin-resistente
Staphylokokken zur parenteralen Verabreichung indiziert. Es
ist dies eine abszedierende Staphylokokkenpneumonie mit Pneumatocele
evtl. mit Pleuraempyem und Spannungspneumatothorax (Abbildung
12). Flucloxacillin wird auch bei der akuten hämatogenen
Osteomyelitis empfohlen.
Abbildung
12: Staphylokokkenpneumonie mit Pneumatocele
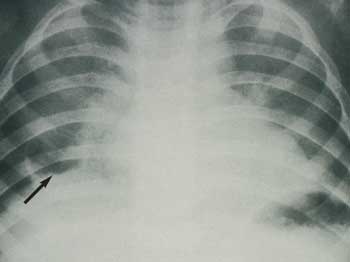
Auch
bei einer abszedierenden Lymphadenitis cervicalis wird das Präparat
allerdings mit mäßigem Erfolg verabreicht. Isoxazolylpenicilline
werden – in Kombination mit einem Aminoglykosid –
zur Behandlung einer Staphylokokken-Endocarditis bzw. bei metastatischen
Absiedelungen aus einem liegenden Fremdkörper verwendet.
Flucloxacillin ist eine therapeutische Option bei schweren Hautaffektionen
z.B. durch Staphylokokken im Neugeborenenalter (Neugeborenen-Pemphigoid)
wegen septischer Metastasen im gesamten Körper, die mit
einer Mortalität von 20% behaftet war (Abbildung 13).
Abbildung
13: Staphylokokkeninfektion der Haut bei einem Neugeborenen
(Neugeborenen-Pemphigoid)
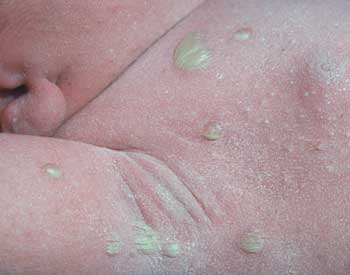
Orale
Isoxazolyl-Präparate haben in der Kinderheilkunde keinen
hohen Stellenwert, da Oralcephalosporine das gleiche Spektrum
abdecken, aber wesentlich besser bioverfügbar sind.
Isoxazolylpenicilline
wirken nicht gegen Methicillin-resistente Staphylokokken (MRSA),
die mit zunehmender Häufigkeit isoliert werden, wobei es
lokale Unterschiede gibt. Vor allem bei Patienten mit Mucoviscidose
sind MRSA häufig, die durch die prophylaktische Gabe von
z.B. Ciprofloxacin induziert werden. Der Resistenzmechanismus
beruht auf der Synthese eines geänderten Penicillin-Bindeproteins.
Bei Koagulase-negativen Staphylokokken ist die Rate an Methicillin-resistenten
Stämmen im Bereich von 60%. Es besteht Kreuzresistenz zwischen
Isoxazolylpenicillinen, Cephalosporinen und Carbapenemen.
Breitspektrumpenicilline
Ampicillin, Amoxicillin und Kombinationen mit Clavulansäure
Eine Änderung der Seitenkette ergab mit erweitertem Wirkspektrum.
Die Aktivität gegen Gram-positive Erreger ist zwar um das
2- bis 5fache niedriger, es werden jedoch zusätzlich eine
Reihe Gram-negativer Mikroorganismen durch Aminopenicilline
erfasst. Es sind dies für die Kinderheilkunde besonders
relevante Erreger wie H. influenzae, Enterokokken,
Listeria monocytogenes. Unterschiedliche Empfindlichkeit
besteht gegen E. coli und Salmonellen, resistent sind
Klebsiella, Enterobacter, Citrobacter, Serratia marcescens,
Pseudomonas und die meisten Proteus-Stämme.
Klinisch
verfügbar sind Ampicillin, Amoxicillin und die Kombination
von Ampicillin mit dem ß-Lactamase-Inhibitor Clavulansäure.
Ampicillin
ist bei oraler Verabreichung nur zu 30–40% bioverfügbar.
Die geringe Bioverfügbarkeit bedingt einerseits niedrige
Serum- und Gewebskonzentrationen. Erreichbare Serumkonzentrationen
liegen bei einer Einzeldosis von 20 mg/kg KG bei 1,5–2
µg/ml. Konzentrationen im Mittelohrsekret liegen abhängig
von der Akuität der Entzündung bei 0,15–0,4
µg/ml Mittelohrsekret. Bei parenteraler Applikation einer
Einzeldosis von 33 mg/kg KG entsprechend einer Tagesdosis von
100 mg/kg KG, aufgeteilt auf 3 Dosen werden 2 Stunden nach Ende
der Kurzinfusion Serumkonzentrationen von 75–120 µg/ml
erreicht. Die Gewebegängigkeit ist gut. Ampicillin geht
in den fetalen Kreislauf und ins Fruchtwasser über.
Ampicillin
wird nach parenteraler Gabe zu 60% unverändert durch die
Niere ausgeschieden, nach oraler Verabreichung zu 20–30%.
Die Wirkstoffkonzentrationen in der Galle sind gleich hoch wie
im Serum.
Nach
oraler Gabe werden bei 5–20%, nach parenteraler Gabe bei
3–10% der Patienten gastrointestinale Nebenwirkungen wie
Übelkeit und Erbrechen, Abdominalkrämpfe und Durchfälle
beobachtet. Es ist jedoch zu betonen, dass ein gewisser Prozentsatz
von Patienten bereits durch die Grundkrankheit zu durchfälligen
Stühlen, z.B. durch Toleranzüberschreitung bei fortgesetzter
Ernährung, neigen und nicht alle Durchfälle antibiotikaassoziiert
sind. Anderseits wird, ähnlich wie nach Gabe von Clindamycin
auch nach parenteraler Verabreichung von Ampicillin eine pseudomembranöse
Enterocolitis beschrieben.
Klinische
Indikation: Wegen der ungenügenden Bioverfügbarkeit
eignet sich Ampicillin vorwiegend zur parenteralen, d.h. intravenösen
Verabreichung. Die Hauptindikationen sind Enterokokkeninfektionen,
wie z.B. eine Enterokokken-Endocarditis in Kombination mit einem
Aminoglykosid oder eine Enterokokken-Harnwegsinfektion. Ampicillin
ist – wiederum in Kombination mit einem Aminoglykosid
– das Mittel der Wahl zur Behandlung einer Listerienmeningitis.
Resorptionsverbesserung
von Aminopenicillinen
Eine Verbesserung der Resorption auf eine Bioverfügbarkeit
von 95% bei Erwachsenen wurde durch einen Resorptionsester (Bacampicillin)
erreicht. Bei Kindern spielte dieses Präparat, nachdem
auch keine Saftform existiert, keine Rolle.
Durch
Hydroxylierung des Ampicillins entsteht das Amoxicillin mit
einer verbesserten Lipoidlöslichkeit, wodurch eine wesentliche
Resorptionsverbesserung erzielt wird. Bei Erwachsenen wird eine
Bioverfügbarkeit von annähernd 100% beschrieben, im
Kindesalter ist die Resorption jedoch erheblich geringer, sodass
bei Kleinkindern zwischen 2 und 5 Jahren eine Bioverfügbarkeit
von 80%, bei Säuglingen im ersten Lebensjahr nur von 65%
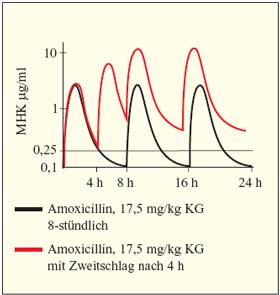
erreicht wird. Dies entspricht bei einer Einzeldosis von 17,5
mg/kg KG einer Serumkonzentration nach 2 Stunden von 5 µg/ml
bei Kleinkindern und von 2,5 µg/ml bei Säuglingen.
Dementsprechend können Konzentrationen im Mittelohrsekret
zwischen 0,5 µg/ml und 1,75 µg/ml erzielt werden.
Nebenwirkungen
der Aminopenicilline
Hautreaktionen sind als häufigste Nebenwirkung bei 5–20%
der Patienten beschrieben. Während eine Allergie bzw. ein
anaphylaktischer Schock nicht häufiger ist als nach Verabreichung
von Penicillin, sind makulöse, hämorrhagische Exantheme
wesentlich häufiger. Besonders hoch ist das Risiko für
ein toxisches Amoxicillin-Exanthem bei einer bestehenden Infektion
durch Epstein-Barr-Viren. Auch ein Erythema exsudativum multiforme
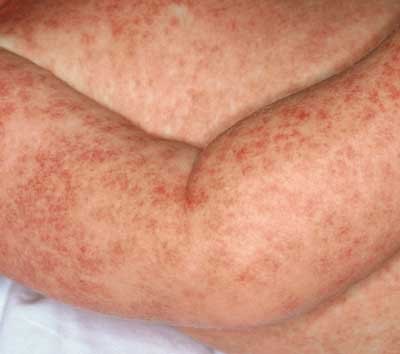
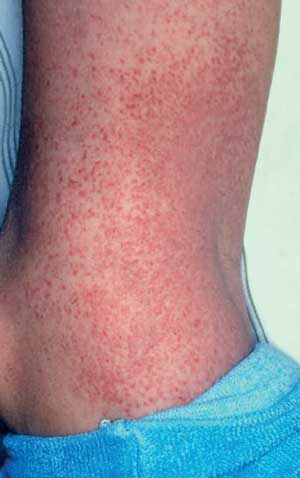
wurde in diesem Zusammenhang beschrieben. Das Exanthem tritt
vorzugsweise am 8.–10. Behandlungstag auf und verschwindet
nach ca. 5–7 Tagen (siehe Abbildung 6a, 6b).
Bei
wiederholter Gabe tritt das Exanthem bereits nach 4–5
Tagen auf. Die Ursache ist unklar. Es wird keine IgE-vermittelte
Überempfindlichkeitsreaktion vom Soforttyp und auch keine
Kreuzallergie mit Penicillin beobachtet. Mehrere Jahre nach
einem typischen Amoxicillinexanthem wird Amoxicillin wiederum
vertragen. Schwere exanthematöse Hauterkrankungen treten
auch bei Gabe von Amoxicillin mit oder ohne Clavulansäure
gehäuft bei der akuten lymphatischen Leukämie auf
(Abbildung 14).
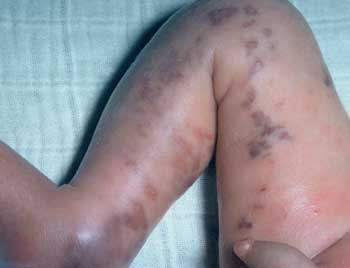
Abbildung
14: Klebsiellen-Sepsis mit Verbrauchskoagulopathie
bei einem Säugling mit Leukämie
Das
antimikrobielle Wirkspektrum und die Nebenwirkungen sind identisch
mit Ampicillin.
Erweiterung
des Wirkspektrums durch Kombination mit ß-Lactamase-Inhibitoren
Sowohl Ampicillin als auch Amoxicillin sind nicht ß-Lactamase-stabil.
Die Kombination mit einem ß-Lactamase-Inhibitor erweitert
das antimikrobielle Spektrum. Clavulansäure wird durch
Fermentation von Streptomyces clavuligerus gewonnen
und ähnelt in der Struktur dem Penicillin-Kern hat aber
keine Azylamino-Seitenkette und in Position 1 anstelle des Sauerstoffmoleküls
Schwefel. Clavulansäure besitzt nur eine schwache antimikrobielle
Eigenwirksamkeit, ist jedoch ein starker irreversibler ß-Lactamase-Hemmer
der Typen II, III, IV und V. Durch die Kombination mit Clavulansäure
ist Amoxicillin gegen ß-Lactamase-bildende Haemophilus-Stämme,
Moraxella catarrhalis, Staphylococcus aureus,
E. coli, Klebsiella pneumoniae, Proteus
spp. und Bacteroides fragilis wirksam. Clavulansäure
schützt Amoxicillin nicht vor Extended-Spectrum-ß-Lactamasen
(ESBL), wie sie von Enterobacter, Serratia,
Morganella, aber auch bestimmten E. coli-Stämmen
gebildet werden.
Es ist zu betonen, dass Amoxicillin in Kombination mit Clavulansäure
gegen Penicillin G-resistente Pneumokokken nicht wirksam ist,
da der Resistenzmechanismus auf einer Änderung der Penicillin-Bindeproteinen
beruht. Auch Methicillin-resistente Staphylokokken werden durch
die Kombination nicht eradiziert.
Die
Kombination Amoxicillin mit Clavulansäure liegt für
pädiatrische Formulierungen in einem bestimmten Mischungsverhältnis
vor. In pädiatrischen Formulierungen ist ein fixes Mengenverhältnis
von 4:1 enthalten, d.h. die Menge an Amoxicillin beträgt
der einer 5 ml-Dosis 125 mg und von Clavulansäure 31,25
mg. Bei Dosissteigerung ist somit das Mischungsverhältnis
immer gleich.
Dies
kann insofern kritisch hinterfragt werden, da die Verträglichkeit
von Clavulansäure erheblich schlechter ist als die von
Amoxicillin. Krampfartige Bauchschmerzen, Übelkeit und
Durchfälle, die zu einer Resorptionsminderung führen,
wurden bei bis zu 25% der Patienten beschrieben. Untersuchungen
zeigten, dass bereits eine kleinere Dosis an Clavulansäure
die ß-Lactamasen irreversibel hemmt und bei einer Dosissteigerung
des antimikrobiell wirksamen Amoxicillin die niedrigere Konzentration
von Clavulansäure und somit ein Mischungsverhältnis
von 6:1 bzw. 8:1 ausreichen würde. Weitere Nebenwirkungen
der Clavulansäure sind ein cholostatischer Ikterus und
eine meist reversible Leberfunktionsstörung. Letale Fälle
wurden in Zusammenhang mit schweren Grunderkrankungen und gleichzeitiger
Medikamentengabe beschrieben.
Die
Kombination von Ampicillin mit Sulbactam verbreitert ebenso
das Spektrum von Aminopenicillinen im Gram-negativen Bereich.
Sulbactam ist ein Penicillansäure-Sulfon mit geringer antibakterieller
Aktivität. Durch die Hemmung der gleichen ß-Lactamasen
ist das Wirkspektrum von Sulbactam-Ampicillin mit dem von Amoxicillin-Clavulansäure
ident. Das orale Präparat enthält einen Ester von
Sulbactam und Ampicillin (Sultamicillin), der im Körper
rasch in die entsprechenden Komponenten gespalten wird. Die
Bioverfügbarkeit ist geringer als die von Amoxicillin ±
Clavulansäure, es werden bei gleicher mg/kg-KG-Dosierung
ca. 25% niedrigere Wirkstoffkonzentrationen als von Amoxicillin
im Serum gefunden.
Bei
intravenöser Verabreichung als Kurzinfusion werden Konzentrationen
im Mischungsverhältnis 1:2 zu Gunsten vom Ampicillin gefunden,
in verschiedenen Geweben kann sich das Mischungsverhältnis
aber erheblich ändern. Gastrointestinale Nebenwirkungen
und Hautreaktionen sind nach Gabe von Sulbactam-Ampicillin seltener
als nach Verabreichung von Amoxicillin-Clavulansäure, auch
Leberschäden sind seltener als nach Gabe von Clavulansäure.
Allergische Reaktionen sind ähnlich selten wie bei Verabreichung
anderer Penicillinderivate. Als Besonderheit wurde nach Gabe
von Sulbactam-Ampicillin jedoch eine – reversible –
Knochenmarksdepression mit Anämie, Thrombopenie und Leukopenie
beschrieben.
Die
Domäne der Aminopenicilline allein oder in Kombination
mit Clavulansäure ist die orale Behandlung von bakteriellen
Infektionen der oberen und unteren Luftwege. Der Großteil
der Infektionen der oberen Luftwege beginnt als Virusinfektion,
die jedoch zu funktionellen Störungen der unspezifischen
Abwehr, z.B. zur Störung der mucociliären Clearance
führen. Daraus resultiert eine bakterielle Superinfektionen
der angrenzenden Strukturen der Nase. Eine bakterielle Superinfektion
der Nasennebenhöhlen ist bei 9–12% der Kleinkinder
und Schulkinder zu beobachten, im Gegensatz zu Erwachsenen,
bei denen bakterielle Superinfektionen bei <2% der Patienten
auftreten. Praktisch jeder akuten Otitis media geht eine Virusinfektion
der oberen Luftwege voraus.
Die
Erreger, mit denen man bei einer akuten Sinusitis konfrontiert
ist, sind Pneumokokken, H. influenzae, Moraxella
catarrhalis in der Reihenfolge ihrer Häufigkeit, seltener,
d.h. in < 5% der Fälle, werden Staphylokokken und Streptokokken
isoliert. Oft besteht eine aerob-anaerobe Mischinfektion z.B.
mit Bacteroides spp.
Bei
einer akuten Otitis media werden die gleichen bakteriellen Mikroorganismen
als Ursache einer Superinfektion in ähnlicher Reihenfolge
bez. ihrer Häufigkeit gefunden. Haemophilus ist
bei Säuglingen und Kleinkindern der häufigste Erreger,
Pneumokokken bei älteren Kindern. ß-Lactamase-bildende
Stämme von Haemophilus influenzae sind in unserem
Einzugsgebiet mit < 10% wesentlich seltener als in den USA
und in Westeuropa, z.B. Spanien, wo zum Teil mehr als 75% der
Isolate ß-Lactamase-Bildner sind. Eigene Untersuchungen
haben gezeigt, dass im ambulanten Bereich die überwiegende
Mehrzahl der primären Isolate bei einer Sinusitis oder
Otitis media auf Amoxicillin empfindlich ist und gleich gute
klinische Ergebnisse mit Amoxicillin wie durch Gabe von Amoxicillin
in Kombination mit Clavulansäure erzielt werden konnten.
Bei einem Rezidiv ist die Situation jedoch anders: Dabei wurden
durch eine antibiotische Vorbehandlung weniger empfindliche
bzw. resistente Mikroorganismen als Ursache einer bakteriellen
Superinfektion selektioniert. Unter diesen Bedingungen kann
auf ein ß-Lactamase-stabiles Breitspektrum-Antibiotikum,
wie z.B. Amoxicillin mit Clavulansäure oder ein Oralcephalosporin
der Cefixim-Gruppe, nicht verzichtet werden.
Eine
klinische Indikation für Amoxicillin allein oder in Kombination
mit einem ß-Lactamaseinhibitor besteht bei der Behandlung
von bakteriellen Infektionen der unteren Luftwege (eitrige Bronchitis,
Bronchopneumonie, Pertussis) vor allem nach einer antibiotischen
Vorbehandlung oder bei chronisch schwelenden Infektionen.
Bei
der antibiotischen Behandlung einer akuten Otitis media ist
zu beachten, dass Ohrschmerz nicht synonym mit einer bakteriellen
Infektion der Paukenhöhle, d.h. einem Paukenhöhlenempyem,
ist. Auch der Unterdruck in der Paukenhöhle als Ausdruck
einer Belüftungsstörung durch die Entzündungsreaktion
im Rahmen der Virusinfektion führt zu Ohrschmerz, Tragusschmerz
und einem gefäß-injizierten Trommelfell. Eine tragbare
Vorgangsweise ist, dass Patienten jünger als 2,5 Jahre
bei Verdacht auf Mittelohrentzündung (unmotiviertes Schreien,
hohes Fieber, Fieberkrämpfe, pathologischer Trom-melfellbefund)
antibiotisch, z.B. mitAmoxicillin oder Amoxicillin+Clavulansäure,
behandelt werden.Bei älteren Kindern kann man mit Fiebersenkung
(Paracetamol, Ibuprofen), antientzündlich (Proxen), abschwellende
Nasentropfen (Oxy-Xylometazolin) mit Verbesserung der mucociliären
Clearance durch Phytopharmaka (Sinupret, 1,8 Cineol) 24–36
Stunden zuwarten, muss den Patienten jedoch dann neuerlich beurteilen
und bei Verschlechterung oder fehlender Besserung mit einem
Aminopenicillin mit oder ohne Clavulansäure oder einem
Oralcephalosprin der Cefixim-Gruppe behandeln.
Harnwegsinfektionen
mit Amoxicillin-resistenten E. coli oder Klebsiellen
sowie Haut- und Weichteilinfektionen mit ß-Lactamase-bildenden
Staphylokokken sind eine weitere klinische Indikation.
Falsche
Indikationen sind eine Monotherapie bei lebensbedrohlichen Infektionen
und eingeschränkter körpereigener Abwehr. Außerdem
sind es gesicherte Streptokokkeninfektionen und Infektionen
durch Clostridien, da hier Penicillin G oder V voll wirksam
sind. Amoxicillin allein und in Kombination mit Clavulansäure
ist bei Mononukleose und akuter lymphatischer Leukämie
kontraindiziert.
Bemerkenswert
ist, dass Isoxazolyl-penicilline auch ß-Lactamasen irreversibel
hemmen. Dadurch kommt auch einer Kombination von Amoxicillin
mit einem Isoxazolylpenicillin ein ähnliches Spektrum zu
wie bei Kombination mit Clavulansäure. Als Präparat
steht Flanamox in einer 1:1-Kombination von Flucloxacillin mit
Amoxicillin zur Verfügung. Da es keine Saftform gibt, wird
dieses Präparat in der Kinderheilkunde nicht eingesetzt.
Azylaminopenicilline
Azylureidopenicilline sind eine Weiterentwicklung des Ampicillins
mit einer Substitution der Aminogruppe durch eine Ureidoseitenkette.
Deshalb sind die Penicillinabkömmlinge dieser Gruppe auch
als Azylureidopenicilline bekannt.
Das
Spektrum dieser Gruppe von Antibiotika umfasst mehr oder weniger
intensiv Pseudomonas aeruginosa, Enterobacterien
und Enterokokken. Alle Präparate sind aber instabil gegen
ß-Lactamasen von Staphylokokken und resistenten Enterobacter-,
Serratia- und Klebsiella-Stämmen.
Präparate
dieser Wirkstoffgruppe sind Azlocillin, Mezlocillin und das
gängigste Piperacillin. Piperacillin hat – wie auch
Mezlocillin – eine gute Wirksamkeit gegen zahlreiche Enterobakterien
und eine dem Azlocillin vergleichbare Wirksamkeit gegen Pseudomonas.
Gegen Enterokokken wirkt Mezlocillin etwas besser. Bei Haemophilus
influenzae und Anaerobiern ist die Wirksamkeit aller Ureidopenicilline
vergleichbar. Azylureidopenicilline sind gegen ß-Lactamase-bildende
Staphylokokken unwirksam und werden durch ß-Lactamasen
von Staphylokokken inaktiviert. Es besteht weitgehend Kreuzresistenz
zwischen den einzelnen Azylaminopenicillinen. Mit Aminoglykosiden
besteht ein Synergismus. Eigene Untersuchungen konnten zeigen,
dass Azylureidopenicilline bei Kombination mit Aminoglykosiden
auch gegen ß-Lactamase-bildende Staphylokokken wirksam
sind, da ein Wirkmechanismus der Aminoglykoside, nämlich
die Hemmung der ribosomalen Proteinsynthese, auch die Bildung
von ß-Lactamasen blockiert. Azylureidopenicilline dürfen
mit Aminoglykosiden nicht gleichzeitig, sondern müssen
zur optimalen Ausnützung der synergistischen Wirksamkeit
sequenziell verabreicht werden.
Die
Präparate sind nur parenteral zur intravenösen Applikation
verfügbar. Die Halbwertszeit aller dieser Medikamente beträgt
1 Stunde, die Plasmaeiweißbindung beträgt 20% für
Piperacillin, 30% für Mezlo- und Azlocillin. Die Gewebegängigkeit
ist gut, die Liquorpenetration ist entsprechend dem Entzündungsgrad
gut bis mäßig.
Azylureidopenicilline
werden zu 60% mit dem Harn ausgeschieden, die Gallekonzentrationen
betragen 30%, ein kleiner Teil wird metabolisiert.
Die
Verträglichkeit der Azylureidopenicilline ist gut, Exantheme
treten seltener als nach Gabe von Ampicillin auf. Gastrointestinale
Symptome sind ebenso selten, ein vorübergehender Anstieg
der Leberenzyme wird bei < 3% der Patienten beobachtet, passagere
Neutropenien sind wie auch nach Gabe anderer ß-Lactam-Antibiotika
möglich.
Das
klinische Einsatzgebiet der Azylureidopenicilline sind schwere
Infektionen der Harnwege, der Gallenwege und des Genitaltraktes
durch empfindliche Enterobakterien. Auch nachgewiesene oder
vermutete Infektionen mit Pseudomonas sind ein Indikationsgebiet.
Auch bei schweren Allgemeininfektionen wie einer Sepsis bei
immunsupprimierten Patienten, einer Meningitis, bei sekundären
Pneumonien bei Patienten mit verschiedenen Grundkrankheiten
wie Herzfehler sind Azylureidopenicilline indiziert. Dabei ist
allerdings die Kombination mit einem Aminoglykosid oder mit
Tazobactam (siehe unten) notwendig, um das Erregerspektrum soweit
als möglich abzudecken.
Eine
Weiterentwicklung des Sulbactams ist das Tazobactam, das als
Kombinationspräparat mit Piperacillin zu einer wesentlichen
Erweiterung des Wirkspektrums beiträgt. Tazobactam, das
selbst keine antimikrobielle Eigenschaft besitzt, hemmt die
meisten Plasmid-übertragbaren ß-Lactamasen und viele
chromosomal codierten Cephalosporinasen der Gruppen I –
IV. Die Kombination von Tazobactam mit Piperacillin ist gegen
die meisten Piperacillin-resistenten ß-Lactamase-bildenden
Stämme einschließlich Pseudomonas aeruginosa
wirksam. Unempfindlich sind Methicillin-resistente Staphylokokken
und ca. 20% der Pseudomonas-Stämme. Enterobacter-,
Serratia- und Klebsiella-Arten sind zu 5 –
10% unempfindlich, Burkholderia cepacia wird nicht
erfasst.
Tazobactam
in Kombination mit Piperacillin ist nur parenteral verfügbar,
die Halbwertszeit beträgt 45 Minuten, die Serumeiweißbindung
ist mit 20% gering. Tazobactam wird zu 75% unverändert
im Harn ausgeschieden. Die Elimination durch die Galle ist gering,
wodurch eine nur mäßige Beeinträchtigung der
Stuhlflora resultiert.
Der
klinische Einsatz von Tazobactam sind schwere, vor allem intraabdominale
Infektionen wie eine Peritonitis, z.B. nach einer perforierten
Appendicitis, Cholangitis, Cholecystitis. Tazobactam wird be
ischweren lebensbedrohlichen Infektionen bei intensivgepflegten
Patienten eingesetzt. Es eignet sich z.B. zur Behandlung einer
nosokomialen Beatmungspneumonie oder nosokomialen Harnwegsinfektion
durch Pseudomonas, Serratien, Acinetobacter.
Zur Sicherheit wird dieses Präparat trotz des erweiterten
Wirkspektrums mit anderen Antibiotika, z.B. Aminoglykosiden,
kombiniert. Tazobactam wird auch bei Tazobactam-empfindlichen
Erregern, insbesondere Pseudomonas aeruginosa oder
Stenotrophomonas maltophilia bei Patienten mit Mucoviscidose
eingesetzt.
Cephalosporine
Eine
wichtige Gruppe innerhalb der ß-Lactam-Antibiotika sind
die Cephalosporine. Der gemeinsame Grundbaustein ist die 7-Aminocephalosporansäure,
die durch unterschiedliche Seitenketten besondere Wirksamkeiten
wie Wirksamkeit gegen Pseudomonas aeruginosa, längere
Halbwertszeit oder überwiegend biliäre Ausscheidung
bedingen.
Der
Wirkmechanismus ist ähnlich wie bei Penicillinen bakterizid
durch Hemmung der Zellwandbiosynthese. Sie wirken auf proliferierende
Mikroorganismen in planktonischem Medium, gegen sessile Keime,
d.h. auf Oberflächen haftende Keime, sind 100–250fach
höhere Wirkstoffkonzentrationen nötig.
Die
Einteilung der Cephalosporine erfolgte früher nach der
historischen Entwicklung in „Generationen“, gegenwärtig
wird eine Einteilung nach Wirkstoffgruppen vorgezogen, die praktikable
Hinweise auf den klinischen Einsatz gibt.
Diese
Gruppen sind:
1)
Cefazolin-Gruppe. Diese Präparate besitzen eine
gute Wirksamkeit gegen Gram-positive Mikroorganismen, einschließlich
ß-Lactamase-bildender Staphylokokken. Gegen Gram-negative
Mikroorganismen ist Cefazolin schwach wirksam. Das Präparat
ist nur parenteral verfügbar. Die Indikationen für
Cefazolin sind durch die Breitspektrum-Cephalosporine stark
eingeschränkt. Cephalosporine der Cefazolin-Gruppe sind
bei Penicillin-Allergie und fehlender Kreuzallergie indiziert.
Cefazolin kann bei leichten Wundinfektionen mit einer ß-Lactamase-bildenden
Mischflora und als perioperative Prophylaxe vor Implantation
von Gelenksprothesen eingesetzt werden. Cefazolin kann auch
prophylaktisch vor Implantation eines PORT-Katheters verabreicht
werden, wenn auch diese Indikationun genügend abgesichert
ist.
2)
Die Cefuroxim-Gruppe besitzt ein intermediäres
Wirkspektrum sowohl gegen Gram-positive als auch gegen zahlreiche
Gram-negative Mikroorganismen, wobei die Wirksamkeit gegen Gram-positive
geringer als bei der Cefazolin-Gruppe und gegen Gram-negative
geringer als bei der Cefotaxim–Gruppe ist. Cefuroxim ist
gegen Ampicillin-resistente Haemophilus-Stämme
wirksam. Zur Cefuroxim-Gruppe gehören das Cefamandol als
erstes Cephalosporin dieser Gruppe, das Cefuroxim und das Cefotiam.
Präparate der Cefuroxim-Gruppe eignen sich wegen des breiten
Wirkspektrum als ungezielte Therapie von Organinfektionen, bei
denen sowohl mit Gram-positiven als auch Gram-negativen Bakterien
gerechnet werden muss. Es sind dies eine ambulant erworbene
Pneumonie und Wundinfektionen. Die Präparate werden bei
schwer kranken Kindern mit Komplikationen von Infektionen der
oberen Luftwege wie einer schweren Sinusitis, mit Orbitalphlegmone,
einer Mastoiditis und einer schweren abszedierenden Lymphadenitis
cervicalis eingesetzt (Abbildung 15, 16, 17).
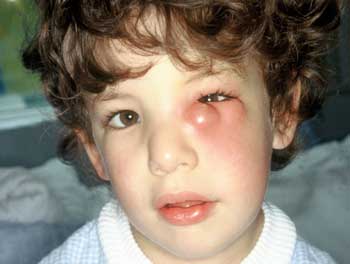
Abbildung
15: Orbitalphlemone/Entzündung des Tränensäckchens
durch Staphylokokken am 3. Tag nach einer Virusrhinitis
Abbildung
16: Idiopathische Wangenphlemone durch Haemophilus
influenzae-Kapsel Typ b
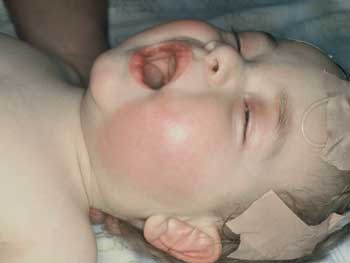
Abbildung
17: Subperiostalabszess bei Mastoiditis
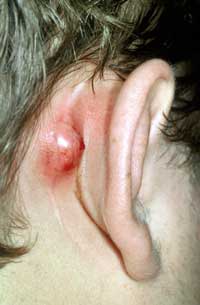
3)
Die Cefotaxim-Gruppe oder Breitspektrum-Cephalosporine
mit besonders guter Wirksamkeit gegen Gram-negative Mikroorganismen.
Die Präparate dieser Gruppe sind Cefotaxim, Ceftriaxon,
Ceftizoxim und Cefmenoxim. Cefotaxim und analoge Antibiotika
wirken besonders gut gegen Klebsiellen, geringer gegen Enterobacter;
Enterokokken sind unempfindlich. Gegen Staphylokokken sind Cepohalosporine
dieser Gruppe deutlich weniger wirksam als Cephalosporine der
Cefazolin- und Cefuroxim-Gruppe. Da der Prozentsatz resistenter
Keime wechseln kann sind zur Schließung von Wirkungslücken
bei schweren lebensbedrohenden Infektionen Antibiotika-Kombinationen
nötig. Cefotaxim ist gegen Streptokokken der Gruppe B gut
wirksam und eignet sich daher zur Ersttherapie bei Früh-
und Neugeboreneninfektionen.
Der
klinische Einsatz von Cephalosporinen der Cefotaxim-Gruppe ist
ähnlich wie bei der Cefuroxim-Gruppe vor allem wenn vermehrt
mit Gram-negativen Erregern gerechnet werden muss. Cefotaxim
eignet sich besonders bei nosokomialen Infektionen bei Früh-
und Neugeborenen, z.B. nach perinataler Asphyxie und Fruchtwasseraspiration,
aber auch zur Behandlung einer Streptokokken B-Sepsis, Meningitis,
Pneumonie.
Cefotaxim
ist das Mittel der Wahl zur Behandlung von Harnwegsinfektionen
in den ersten Lebensjahren, die wegen der Schwere des Krankheitsbildes
und der oft begleitenden Durchfällen intravenös behandelt
werden müssen.
Cefotaxim
bzw. Ceftriaxon sind gegenwärtig das Mittel der Wahl zur
Behandlung der eitrigen Meningitis durch Meningokokken, Pneumokokken
und dem sehr selten gewordenen Haemophilus influenzae.
Dabei ist eine Tagesdosis von 200 mg/kg KG bei Cefotaxim auf
3 Dosen bzw. 100 mg/kg KG bei Ceftriaxon am 1. Tag aufgeteilt
auf 2 Dosen nötig. Die erste Dosis muss als Kurzinfusion
über 2–3 Stunden verabreicht werden. Cefotaxim eignet
sich nicht zur Behandlung einer Ventilinfektion bzw. einer Infektion
einer externen Ventrikeldrainage.
Ceftriaxon
wird vorwiegend bei der Borrelienmeningitis, aber auch zur Behandlung
einer Borellienarthritis eingesetzt, da es wegen der Einmalgabe
auch ambulant verabreicht werden kann. Die Dosierung beträgt
75–100 mg/kg KG.
Cefotaxim
ist der Kombinationspartner entweder mit einem Aminoglykosid
oder mit einem Glykopeptid bei der ungezielten Behandlung schwerer
bakterieller Infektionen bei immunsupprimierten Patienten, z.B.in
der Akutphase einer Leukämiebehandlung (Abbildung 18).
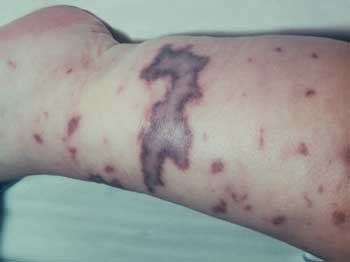
Abbildung
18: Meningokokkensepsis
Falsche
Indikationen sind leichte bakterielle Infektionen, bei denen
ebenso mit Penicillin, Aminopenicillinen oder einem Oralcephalosporin
der Cefixim-Gruppe behandelt werden kann.
4)
Die Ceftazidim-Gruppe (oder Pseudomonas-Cephalosporine)
besteht aus Ceftazidim, Cefepim und Cefpirom und besitzt eine
besondere Pseudomonas-Wirksamkeit. Ceftazidim hat fast das gleiche
Wirkspektrum wie Cefotaxim, ist aber gegen Pseudomonas aeruginosa
10-mal stärker wirksam als Cefotaxim. Ceftazidim besitzt
auch gegen Enterobacter, Serratia und Acinetobacter
eine gute Wirksamkeit, gegen Staphylokokken ist das Präparat
3fach schwächer als das ohnehin schwache Cefotaxim. Ceftazidim
ist gegen Methicillin-resistente Staphylokokken, Enterokokken,
Listerien und Clostridium difficile unempfindlich.
Die Präparate dieser Gruppe haben eine Serum-Halbwertszeit
von 2 Stunden und eine Serum-Eiweißbindung von 10% (Ceftazidim)
und 20% (Cefepim). Die Gewebspenetration ist gut, ebenso die
Liquorpenetration, jedoch wiederum abhängig vom Entzündungsgrad.
Ceftazidim-resistente Pseudomonas-Stämme können
gegen Cefepim empfindlich sein. Die Pseudomonas-Wirksamkeit
von Cefpirom kann lokal unterschiedlich schwächer sein,
die Wirksamkeit gegen Enterobacter ist oft stärker,
gegen Acinetobacter und Serratia, aber auch
gegen Staphylokokken ähnlich oder stärker als von
Cefotaxim.
Die
Indikation für Ceftazidim ist die gezielte Behandlung von
Pseudomonas-Infektionen vorwiegend in Kombination mit
einem Aminoglykosid. Ceftazidim ist auch als Interventionstherapie
in Kombination mit einem Aminoglykosid bei neutropenischen Patienten
indiziert. Die Verabreichung der Kombinationspartner sollte
nicht gleichzeitig, sondern um 4–6 Stunden zeitversetzt
erfolgen. Wenn bei einer ungezielten Therapie einer schweren
Infektion bei eingeschränkter körpereigener Abwehr
eine Beteiligung von Staphylokokken oder Anaerobiern nicht ausgeschlossen
werden kann, ist eine Kombination mit Clindamycin oder einem
Glykopeptid möglich.
Eine
wichtige Indikation ist die Behandlung von Pseudomonas-Infektionen
bei der Mucoviscidose.
5) Die Cefoxitin-Gruppe oder Anaerobier-Cephalosporine.
Zu dieser Gruppe gehören Cefoxitin und Cefotetan. Gemeinsam
ist diesen Cephalosporinen die hochgradige ß-Lactamase-Stabilität
einschließlich der ß-Lactamasen, die von Bacteroides
fragilis gebildet werden. Deshalb ist eine sehr gute Wirksamkeit
gegen Anaerobier gegeben. Resistent sind Pseudomonas
und Enterokokken sowie Methicillin-resistente Staphylokokken,
Enterobacter und Citrobacter. Gegen Haemophilus
ist die Wirksamkeit geringer als die der Cephalosporine der
Cefuroxim- und Cefotaxim-Gruppe.
Das
klinische Indikationsgebiet umfasst vor allem die ungezielte
Therapie von Infektionen, bei denen mit Anaerobiern bzw. einer
aerob/anaeroben Mischflora zu rechnen ist. Es sind dies abszedierende
Pneumonien, z.B. nach Fremdkörperaspiration, eine Mundbodenphlegmone
oder ein Tonsillarabszess. Zur Steigerung der Sicherheit ist
die Kombination mit einem Aminoglykosid sinnvoll.
Cefoxitin
wird auch gezielt zur Behandlung von Infektionen mit sensiblen
Mikroorganismen, die gegen andere Cephalosporine unempfindlich
sind, verwendet.
6)
Oralcephalosporine der Cefalexin-Gruppe besitzen
ein ähnliches Wirkspektrum wie die Cephalosporine der Cefazolin-Gruppe
mit guter Wirksamkeit gegen Gram-positive Mikroorganismen und
einer relativ bescheidenen Wirksamkeit gegen Gram-negative Bakterien.
Sie zeichnen sich durch eine günstige Pharmakokinetik aus
und eine nahezu komplette Bioverfügbarkeit. Cephalosporine
werden unabhängig von einer gleichzeitigen Nahrungszufuhr
resorbiert. Serumkonzentrationen sind bei einer Einzeldosis
von 20 mg/kg KG Cefalexin, Cefadroxil 5-mal höher als nach
einer äquimolaren Gabe von Amoxicillin.
Cefalexin
war das erste Oralcephalosporin mit Aktivität gegen Staphylokokken
einschließlich ß-Lactamase-bildender Stämme,
jedoch mit Ausschluss der MRSA. Cefalexin ist auch gut gegen
Streptokokken, E. coli, Proteus mirabilis
und Klebsiella spp. wirksam, gegen Streptokokken ist
die Wirksamkeit jedoch um das 10fache schwächer als von
Penicillin. Unempfindlich sind Enterokokken und Haemophilus
influenzae.
Cefadroxil
ist in seinem Wirkspektrum dem Cefalexin ähnlich. Es hat
bei einer nahezu kompletten Bioverfügbarkeit eine Halbwertszeit
von 1,5 Stunden und kann damit in 2 Tagesdosen verabreicht werden.
Cefaclor
wirkt auf Streptokokken und Pneumokokken sowie auf empfindliche
Gram-negative Enterobakterien 4- bis 8mal stärker als die
übrigen Oralcephalosporine, zudem hemmt Cefaclor auch Amoxicillin-empfindliche
und Amoxicillin-resistente Haemophilus influenzae-Stämme.
Die Bioverfügbarkeit von Cefaclor beträgt 80%. 60%
wird im Harn unverändert ausgeschieden, ein Teil wird im
Körper metabolisiert. Die Resorption von Cefaclor wird
individuell unterschiedlich durch gleichzeitige Nahrungsaufnahme
beeinflusst, es kommt insbesondere bei Säuglingen zu einer
Resorptionseinbuße von bis zu 50%. Die Rate an Nebenwirkungen
ist bei Gabe von Cefaclor gering, das Präparat führt
in < 3% der Patienten zu gastrointestinalen Nebenwirkungen.
Eine vorübergehende Erhöhung von SGOT und SGPT sowie
der alkalischen Phosphatase wurde beschrieben. Bemerkenswert
ist der gute Geschmack von Cefaclor, der die Compliance erheblich
erleichtert.
Loracarbef
ist ein synthetisches ß-Lactam-Antibiotikum der Carbacephem-Klasse
und wird ausschließlich oral verabreicht. Die antimikrobielle
Wirksamkeit entspricht der von Cefaclor, das Präparat ist
jedoch gegen Haemophilus influenzae, Moraxella
catarrhalis und einigen Enterobacteriaceae, besonders
gegen ß-Lactamase-bildende Stämme, stärker wirksam.
Die Bioverfügbarkeit von Loracarbef ist nahezu vollständig,
nach einer Einzeldosis von 20 mg/kg KG werden Konzentrationen
im Serum von 30 µg/ml erreicht, die Halbwertszeit beträgt
1 Stunde. Die Gewebegängigkeit ist ebenso sehr gut. Im
Mittelohrsekret betragen die Wirkstoffkonzentrationen nach 2
Stunden ca. die Hälfte der gleichzeitig gemessenen Serumkonzentrationen
von 12–15 µg. Das Präparat wird zu 90% unverändert
im Harn ausgeschieden, es findet praktisch keine Metabolisierung
statt. Dies hat zur Folge, dass im Gegensatz zu Cefaclor die
Stuhlflora weitgehend unbeeinflusst bleibt.
Die
Verträglichkeit von Loracarbef ist gut, selten treten Bauchschmerzen,
Übelkeit, Erbrechen und Durchfälle auf. Hautrötungen
und Exantheme sind selten. Ein serum-krankheitsähnliches
Erscheinungsbild mit Ödemen, Gelenkschwellung, Fieber und
Hautreaktion wurde beobachtet.
Der
klinische Einsatzbereich sind vor allem Infektionen der Luftwege
mit empfindlichen Keimen, insbesondere Staphylokokken. Cephalosporine
der Cefalexin-Gruppe können als Alternative auch zur Behandlung
einer Streptokokkenangina, z.B. bei Penicillinallergie und fehlender
Kreuzallergie, verwendet werden, bieten aber sonst keinen Vorteil.
Diese Cephalosporine eignen sich auch zur Behandlung von Haut-
und Weichteilinfektionen mit einer ß-Lactamase-bildenden
Mischflora, z.B. nach einem Hundebiss oder bei großen
infizierten Schürfwunden.
7)
Oralcephalosporine mit erweitertem Spektrum (Oralcephalosporine
III)
Zu dieser Gruppe gehören neben Cefixim (Cephoral, Suprax)
und Ceftibuten Prodrugs wie Cefuroxim-Axetil, Cefpodoxim-Proxetil,
Cefetamet-Pivotil, die in der Darmwand hydrolysiert werden und
nach der Resorption freie Wirkstoffkonzentrationen der einzelnen
Präparate im Serum aufscheinen.
Cefixim
weist generell eine erheblich stärkere In vitro-Aktivität
als Cefalexin und Cefaclor auf. Im Vergleich zu diesen Antibiotika
weist Cefixim gegen Haemophilus influenzae eine 6fach
stärkere, gegen Streptococcus pyogenes eine 10fach
stärkere und gegen Klebsiella und Proteus
spp eine 30- bis 50fach stärkere antimikrobielle Wirksamkeit
auf. Gegen Enterobacter cloacae, Proteus vulgaris
und Morganella morganii ist Cefixim wirksam, während
Cefalexin und Cefaclor unwirksam sind. Gegen Pneumokokken ist
die Wirksamkeit gleich, gegen Staphylokokken etwas schwächer.
Resistent sind MRSA, Penicillin-resistente Pneumokokken, Enterokokken,
Pseudomonas aeruginosa, Clostridium difficile
und Bacteroides fragilis. Natürlich sind auch
intrazelluläre Mikroorganismen wie Mykoplasmen, Chlamydien
unempfindlich.
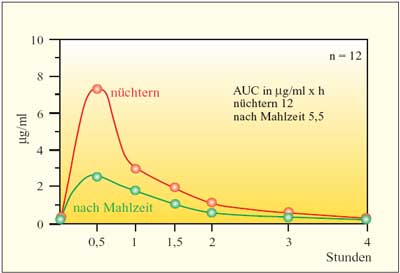
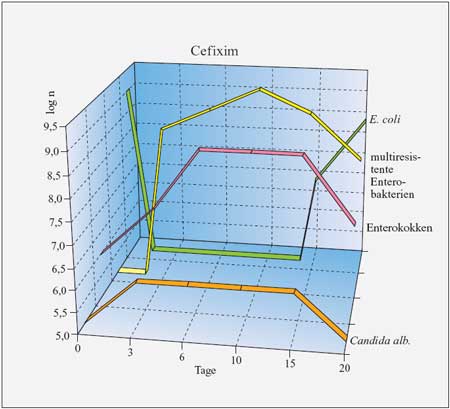
Cefixim
zeigt als erster Vertreter dieser Gruppe nur eine ca. 50%ige
Bioverfügbarkeit. Nach oraler Gabe von 5 mg/kg KG liegen
die Serumkonzentrationen nach 3 Stunden bei 1,5 µg/ml.
Die Halbwertszeit beträgt 2,5 Stunden, die Eiweißbindung
65%. Das Präparat wird nur zu 20% renal eliminiert, es
finden sich hohe Gallenkonzentrationen. Dies und die unvollständige
Resorption führen zu einer erheblichen Störung der
Homöostase der Darm- bzw. Stuhlflora und Überwucherung
mit Enterokokken, Candida spp und Selektion multiresistenter
Enterobakterien.
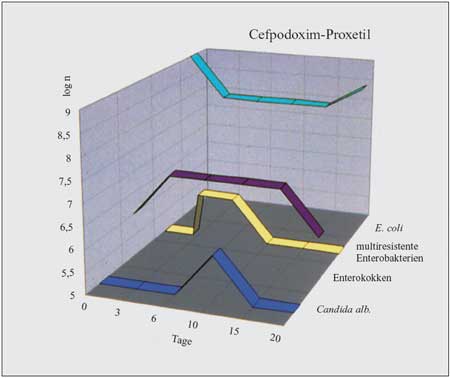
Cefpodoxim-Proxetil
ist ein Resorptionsester des Ceftizoxims, der in der Darmwand
vollständig zu aktiven Cefpodoxim hydrolysiert wird. Cefpodoxim
ist gegen Streptokokken und Pneumokokken 10-mal wirksamer als
Cefaclor, gegen Haemophilus 30-mal wirksamer. Staphylokokken
und Moraxella catarrhalis sind gleich gut empfindlich.
Cefpodoxim ist gegen Gram-negative Bakterien wesentlich wirksamer
als Cefaclor. Unempfindlich sind Pseudomonas aeruginosa,
Serratia marcescens, Enterokokken und Bakteroides-Arten
sowie Methicillin-resistente Staphylokokken.
Cefpodoxim
wird ebenso nicht vollständig resorbiert. Nach Nahrung
wird Cefpodoxim etwas besser resorbiert als nüchtern. Maximale
Serumkonzentrationen 2 Stunden nach einer Einmalgabe von 5 mg/kg
KG betragen 3,5 µg/ml, die Halbwertszeit beträgt
2,5 Stunden. Der Verdacht, dass durch die Unreife der Enzymausstattung
der Resorptionsester bei Säuglingen im ersten und zweiten
Trimenon schlechter resorbiert würde, konnte nicht bestätigt
werden. Das Präparat wird überwiegend renal eliminiert,
die Gallenkonzentrationen betragen 3–4 µg/ml. Dadurch,
dass das Präparat im Darm nicht antimikrobiell wirksam
ist, und die niedrigen Wirkstoffkonzentrationen in der Galle
führen zu einer geringen Störung der Stuhlflora, resistente
Enterobakterien, Enterokokken und Candida albicans
werden kaum selektioniert. Durchfälle kommen gelegentlich,
allergische Erscheinungen selten vor.
Cefuroxim-Axetil
ist der Azetoxyäthylester von Cefuroxim, der in der Darmwand
hydrolysiert wird, wodurch 2 Moleküle freies Cefuroxim
im Blut erscheinen. Die Wirksamkeit von Cefuroxim ist unter
der Gruppe 2 beschrieben. Das Präparat ist weitgehend ß-Lactamase-stabil
und wirkt gegen Pneumokokken, Streptokokken und auch gegen Ampicillin-resistente
Haemophilus und Penicillin-resistente Pneumokokken.
Auch im Gram-negativen Bereich ist Cefuroxim stärker wirksam
als Cefaclor und Cefalexin. Insgesamt ist die Wirksamkeit aber
zwischen der von Cefaclor und der von Cefpodoxim-Proxetil anzusiedeln.
Resistent sind Pseudomonas aeruginosa, Enterobacter
und MRSA.
Auch
bei Cefuroxim-Axetil ist die Resorption unvollständig.
Die im Harn wiederentdeckte Wirkstoffmenge beträgt 30–40%,
es besteht eine nennenswerte biliäre Exkretion mit Nachweis
von Wirkstoffkonzentration im Intestinalsekret und ein Selektionsdruck
auf die Stuhlflora.
Cefetamet
ist ein Cefotaxim-Derivat, das als Pivaloyl-Oxymethylester nach
oraler Gabe resorbiert wird. Das Präparat ist um den Faktor
8–10 stärker wirksam gegen Streptococcus pyogenes,
Haemophilus influenzae, E. coli, Klebsiella und
Yersinien. Die Bioverfügbarkeit ist gut, die im Harn ausgeschiedene
Wirkstoffmenge beträgt 50%. Die Halbwertszeit liegt bei
3 Stunden, was eine Einmalgabe ermöglicht. Da keine Saftform
existiert, ist dieses Präparat in der Kinderheilkunde nicht
eingeführt.
Ceftibuten
besitzt ein ähnliches Wirkspektrum wie Cefixim und ist
gegen Plasmide- und Chromosomal-vermittelte Plasmiden stabil.
Es ist gegen Streptokokken und Haemophilus wirksam,
aber gegen Pneumokokken, vor allem aber gegen Staphylokokken
weniger wirksam als Cefpodoxim. Der Vorteil von Ceftibuten besteht
in der guten Aktivität gegen zahlreiche Enterobacteriaceae.
Enterobacter und Serratia sind häufig,
Pseudomonas aeruginosa ist immer resistent.
Die
Bioverfügbarkeit von Ceftibuten ist gut, die im Harn ausgeschiedene
Wirkstoffmenge beträgt 60–70%. Damit eignet sich
dieses Präparat zur Behandlung von Harnwegsinfektionen
bei Kleinkindern, bei denen eine orale Behandlung möglich
erscheint.
Der
klinische Einsatz von Oralcephalosporinen III der Cefixim-Gruppe
ist breit. Diese Präparate geben eine hervorragende Behandlungssicherheit
bei Otitis media, Sinusitis, ambulant erworbener Pneumonie.
Auch eine Streptokokkenangina kann bei Penicillin-Überempfindlichkeit
mit einem dieser Präparate behandelt werden.
Diese
Cephalosporine eignen sich auch sehr gut zur ambulanten Behandlung
von schweren Harnwegsinfektionen. Bei Harnwegsfehlbildungen
können diese Präparate auch zur Dauerprophylaxe verwendet
werden, wenn bereits resistente Mikroorganismen selektioniert
wurden. Durch Cefixim wird jedoch regelhaft ein multiresistenter
Enterobacter cloacae oder Candida spp. als
Reinfektionserreger selektioniert.
Das
am besten geeignete Präparat aus dieser Gruppe ist das
Cefpodoxim-Proxetil, das vor allem wegen der günstigen
Pharmakokinetik und Pharmakodynamik sehr gute klinische Ergebnisse
erbringt, vielfach den Übergang in eine chronisch schwelende
Infektion verhindert und die körpereigene Flora am geringsten
von allen Breitspektrum-Cephalosporinen beeinträchtigt.
|