| Quantifizierung von PCR-Produktmengen durch real-time PCR-Verfahren |
|
D. Wilfingseder, H. Stoiber |
| Schlüsselwörter:
PCR,quantitativereal-timePCR,SYBR®-Green I, TaqMan®-Prinzip, CT-Wert |
Zusammenfassung Die real-time quantitative PCR stellt eine moderne Technologie dar, die im molekularbiologischen Bereich sehr breite Verwendung findet. Im Vergleich zur Endpunkt-PCR, bei der DNA-Produkte qualitativ nachgewiesen werden können, wurde es durch Entwicklung der quantitativen real-time PCR möglich, Aussagen über die Ausgangs-DNA-Mengen zu machen. Dies macht die real-time quantitative PCR zu einem unentbehrlichen Werkzeug sowohl bei Sequenzanalysen, Genklonierungs- und Genexpressionsstudien, als auch im diagnostischen Bereich zum Nachweis von verschiedenen Pathogenen. |
Key-words: PCR,quantitativereal-timePCR,SYBR®-Green I, TaqMan®-principle, CT-value |
Summary Quantitative real-time PCR is a modern technology widely applied in the field of molecular biology. When compared to endpoint PCR, which allows only qualitative analyses of generated DNA-products, quantitative PCR allows highly accurate measurement of DNA-amounts and therefore quantitation of initial amounts of DNA. This feature makes real-time quantitative PCR a valuable tool in a variety of applications like sequence analyses, gene cloning and gene expression studies or determination of different pathogens. |
| Definition
Die real-time quantitative PCR oder
auch quantitative Echtzeit-PCR stellt eine Weiterentwicklung der
Mitte der achtziger Jahre von K.B. Mullis entwickelten Polymerase-Kettenreaktion
(polymerase chain reaction, PCR) dar [1]. Die PCR ist eine sehr
schnelle und sensitive Methode zur In-vitro-Amplifizierung
spezifischer DNA-Abschnitte und ermöglicht somit die Detektion
kleinster DNA-Mengen. Die PCR ist ein sehr wichtiges Werkzeug der
modernen Molekularbiologie geworden, da sie im Bereich der Sequenzanalyse,
bei Genexpressionsstudien und bei der Genklonierung eingesetzt wird
[2, 3]. Das Prinzip der PCR-Reaktion basiert auf der enzymatischen
Vermehrung eines bestimmten DNA-Abschnittes, der zwischen zwei Startersequenzen,
den so genannten Primern, liegt. Die Basenabfolge der beiden Primer
muss komplementär zur amplifizierenden DNA-Sequenz sein. Weiters
sollten die Primer eine Länge von 18 bis 30 Basen haben, einen
G/C-Gehalt zwischen 20-80%, sowie eine Schmelztemperatur (Tm) von
ca. 60°C. Bei der Primerauswahl sollten Poly(T)-Bereiche, Haarnadelstrukturen
und auch |
| Real-time
PCR
Die Grundlage für die heute angewandte real-time quantitative PCR wurde 1992 durch Higuchi et al. geschaffen [5]. Higuchi et al. statteten die PCR-Maschine mit einer UV-Lampe und CCD-Kamera aus und fügten der PCR-Reaktion Ethidiumbromid (EtBr) zu. EtBr fluoresziert, wenn es in doppelsträngige DNA eingebaut und durch UV-Licht angeregt wird. So konnte die Fluoreszenz gemessen und die Konzentration der Ziel-DNA bestimmt werden [6]. Die Methode wird auch heute noch angewandt, jedoch unter Verwendung anderer Farbstoffe. Bei der real-time PCR wird ein fluoreszierender Reporterfarbstoff verwendet, um die Reaktion verfolgen zu können. Dieser Reporter kann von nicht-spezifischer Natur sein oder spezifisch mit der Ziel-DNA interagieren. In beiden Fällen steigt die Fluoreszenz proportional mit der Produktmenge an. Real-time Detektionssysteme bestehen im Prinzip aus einem PCR-Cycler sowie einem optischen Detektionsmodul, über das die mit der Produktzunahme ansteigenden Fluoreszenzwerte online nach jedem Zyklus gemessen werden. Die Auswertung und Quantifizierung erfolgt mittels der geeigneten Computersoftware. Die Fluorophore werden – je nach System – mit Halogen-, LED- oder Laserlicht angeregt. Die Quantifizierung der PCR basiert
bei allen Systemen auf der Berechnung des Fluoreszenz-Schwellenwertes,
dem so genannten Threshold Cycle oder CT-Wert.
Der CT-Wert ist jener PCR-Zyklus, bei dem
die Reporterfluoreszenz die Hintergrundfluoreszenz signifikant übersteigt.
Am Anfang der PCR-Reaktion wird nur die Basis- oder Hintergrundfluoreszenz
gemessen, da die Reporterfluoreszenz aufgrund der geringen Templatekonzentration
im Reaktionsgefäß während der ersten PCR-Zyklen
normalerweise nicht detektierbar ist. Die Quantifizierung der DNA-Menge
beruht nicht auf absoluten Mengen an PCR-Produkt, sondern auf der
Kinetik der PCR-Reaktion. Dafür nimmt man als Richtlinie den
CT-Wert, da zu diesem Zeitpunkt die Amplifikation
exponentiell ist und es in dieser Phase der PCR-Reaktion keine limitierenden
Faktoren, wie Primer- oder Nucleotidmangel, nachlassende Enzymaktivität
oder Inhibition der PCR-Reaktion durch Generation bestimmter Produkte,
gibt. Parallel dazu werden in jedem PCR-Lauf bekannte Templatemengen
amplifiziert, sodass man vergleichen kann, welche Templatemenge
man bei welchem CT-Wert erhält. Daraus
lässt sich eine Standardkurve erstellen, anhand derer man aus
einem bestimmten CT-Wert auf eine Templatekonzentration
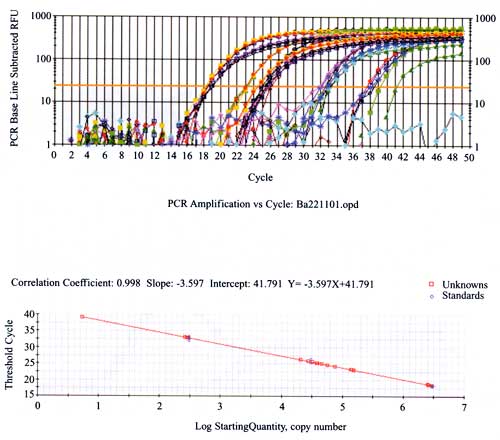
schließen kann. Ein typischer real-time PCR-Run und die daraus
resultierende Standardkurve ist in Abb. 1 dargestellt.
|
| Real-time
PCR und interkalierende Farbstoffe
Wie bereits erwähnt, verwendet
man heutzutage andere interkalierende Farbstoffe als EtBr, die ein
besseres Signal-Hintergrund-Verhältnis liefern, hier vor allem
SYBR®-Green I (Molecular Probes, Portland, Oregon). Dieser Fluoreszenzfarbstoff
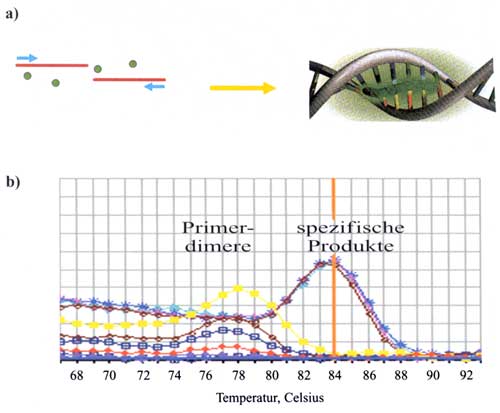
lagert sich – wie EtBr – unspezifisch in Doppelstrang-DNA
ein (Abb. 2a). Dadurch kommt es mit fortschreitender PCR-Reaktion
zu einem Fluoreszenzanstieg. Der Vorteil von SYBR®-Green I ist
die universelle Verwendbarkeit, da es unspezifisch eingebaut wird
und in jede beliebige PCR-Reaktion eingesetzt werden kann, sowie
die hohe Signalstärke, da jedes DNA-Molekül mehrere Fluoreszenzmoleküle
bindet. Es fehlt jedoch eine spezifische Bindung des Fluorophors
an die zu amplifizierende Ziel-DNA, sodass eine Unterscheidung zwischen
korrektem Produkt und Artefakt oder Primerdimeren, die während
der PCR-Reaktion auch einen Fluoreszenzanstieg verursachen können,
nicht möglich ist. Eine Differenzierung zwischen spezifischem
Produkt und Primerdimeren beziehungsweise auch Mutationsanalysen
sind im Anschluss an den PCR-Run mithilfe einer Schmelzkurvenanalyse
möglich. Dabei kommt es durch schrittweisen Temperaturanstieg
zu einer Auftrennung der DNA-Doppelstränge entsprechend ihrer
jeweiligen Schmelzpunkte in ihre Einzelstränge. Die daraus
resultierende Fluoreszenzabnahme wird aufgezeichnet. Aufgrund der
Schmelztemperaturen kann man zwischen spezifischen Produkten und
Primerdimeren oder Mutationen unterscheiden, da Primerdimere/Mu-tationen
bei geringeren Temperaturen schmelzen als die spezifischen, größeren
PCR-Produkte (Abb. 2b).
|
| Spezifische
Hybridisierungssonden
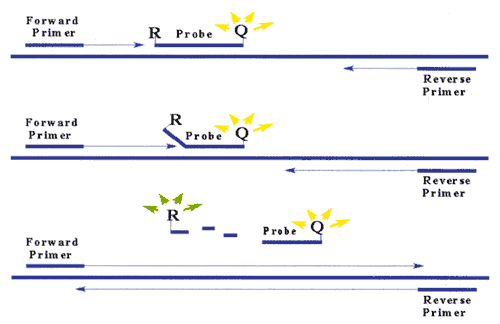
Das Problem der mangelnden Spezifität bei der Verwendung von interkalierenden Farbstoffen konnte durch Design von spezifischen Hybridisierungssonden gelöst werden. TaqMan-Sonden
Die Zunahme der Reporterfluoreszenz
wird nach jedem Zyklus gemessen und ist wiederum proportional der
Menge des DNA-Templates im Tube. Die Taq-Polymerase fragmentiert
nur an die Zielsequenz gebundene TaqMan-Sonden, nicht hybridisierte
Einzelstränge bleiben unbeschadet. Häufig wird Fluorescein
(FAM) als Reporter und Rhodamin (TAMRA) als Quencher verwendet,
für eine real-time quantitative Multiplex-PCR, bei der mehrere
Zielsequenzen in einer einzelnen PCR- Reaktion nachgewiesen werden,
können Farbstoffe mit unterschiedlichen Extinktions- und Emissionswellenlängen
als Reporter für die nachzuweisenden Sequenzen hinzugenommen
werden (Tab. 1). Anstelle von TAMRA kann auch ein „Dark Quencher“
(Dabcyl, Methylorange) genommen werden, der den Wellenlängenbereich
um 585 nm für weitere Reporterfarbstoffe freigibt. Ein Dark
Quencher kann im Vergleich zu TAMRA die Lichtemission des Reporterfluorophors
noch besser unterdrücken, interferiert auch nicht mit der Messung
und wird vor allem für die Multiplex-PCR wie auch die Markierung
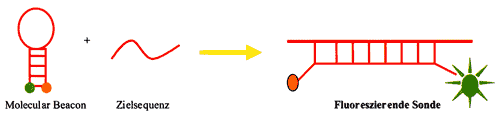
von Molecular Beacons eingesetzt.
MGB-Proben® |
| Ausblick
Die real-time quantitative PCR stellt
ein relativ neues, grundlegendes Werkzeug der modernen Molekularbiologie
dar und erlaubt Sequenz- und Mutationsanalysen, Genexpressions-
und Genklonierungsstudien, Überprüfung des Virustiters
unter Therapie sowie den diagnostischen Nachweis von Bakterien und
Viren in kürzester Zeit und mit höchster Reproduzierbarkeit.
Bei der real-time quantitativen PCR handelt es sich um eine sehr
sensitive Methode, die Messungen über einen Bereich von 7 bis
8 log-Stufen und den Nachweis von kleinsten DNA-Kopienmengen zulässt.
Im Bereich der real-time quantitativen PCR findet eine rasante Entwicklung
statt, und Berichte, Publikationen oder Protokolle über die
Einsatzmöglichkeiten der real-time quantitativen PCR häufen
sich täglich. Zurzeit bieten sechs verschiedene Hersteller
(ABI PRISM®-5700SDS, 7700SDS, 7900HT-SDS und 7000SDS von PE-Applied
Biosystems, Light Cycler® von Roche-Diagnostics, iCycler®
von BioRad, Mx4000® von Stratagene, Smart Cycler® System
von Cepheid, Rotorgene® von Corbett Research) real-time PCR-Geräte
an, es werden jedoch sicher weitere folgen. Die Geräte unterscheiden
sich unter anderem bezüglich ihrer Bauweise, der Lichtquellen,
mit denen die Fluorophore angeregt werden, und der Durchsatzraten.
Von den meisten Herstellern der real-time quantitativen PCR-Geräte
werden auch dazugehörige Kits und Reagenzien und allgemeine
Richtlinien für die Reaktionsbedingungen angeboten, es ist
bei einigen Geräten aber auch möglich, so genannte in-house
Mixes zu verwenden. Natürlich kann mit den real-time PCR-Geräten
auch Reverse Transcription-PCR (real-time quantitative RT-PCR) durchgeführt
werden, wenn vor der PCR ein reverser Transkriptionsschritt programmiert
wird. Zwei Beispiele für ein typisches real-time quantitative
RT-PCR-Protokoll und Thermoprotokoll sind in Tabelle 2 dargestellt
(AB-Kit, Stratagene-Kit). Somit stellt die real-time quantitative
PCR eine relativ einfache Methode, mit der man schnell sehr präzise
Ergebnisse erzielen kann, dar.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Literatur:
1. Mullis K.B., Faloona F.A.: „Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction.“ Methods Enzymol. 155 (1987) 335-50. 2. Scharf S.J., Horn G.T., Erlich H.A.: „Direct cloning and sequence analysis of enzymatically amplified genomic sequences.“ Science 233 (1986) 1076-8. 3. Erlich H.A., Gelfand D., Sninsky J.J.: „Recent advances in the polymerase chain reaction.“ Science 252 (1991) 1643-51. 4. Gilliland G., Perrin S., Blanchard K., Bunn H.F.: „Analysis of cytokine mRNA and DNA: detection and quantitation by competitive polymerase chain reaction.“ Proc. Natl. Acad. Sci. USA 87 (1990) 2725-9. 5. Higuchi R., Dollinger G., Walsh P.S., Griffith R.: „Simultaneous amplification and detection of specific DNA sequences.“ Biotechnology (N Y) 10 (1992) 413-7. 6. Higuchi R., Fockler C., Dollinger G., Watson R.: „Kinetic PCR analysis: real-time monitoring of DNA amplification reactions.“ Biotechnology (N Y) 11 (1993) 1026-30. |
| Anschrift
des Verfassers: A. Univ.-Prof. Dr. Heribert Stoiber Institut für Hygiene und Sozialmedizin der Universität Innsbruck A-6020 Innsbruck, Fritz-Pregl-Straße 3 E-Mail: heribert.stoiber@uibk.ac.at |