| Ketolide Eine neue Antibiotikaklasse |
|
|
|
W. Graninger |
|
Ketolide sind eine neue Antibiotikaklasse, die aus der Erythromycin-Familie hervorgegangen sind. Sie besitzen einen makrozyklischen Lactonring, der an den Seitenketten wesentliche Veränderungen aufweist. Der Makrolid-typische Zucker, die Cladinose, an Position C3 ist durch eine neuartige Keto-Gruppe ersetzt. Diese Keto-Gruppe an Position 3 des 14-gliedrigen Lactonringes hat der Klasse ihren Namen gegeben. Die Keto-Gruppe trägt entscheidend dazu bei, dass die Ketolide eine außerordentlich hohe Säurestabilität aufweisen. Selbst bei pH 1 ist die Substanz nach 6 Stunden noch zu 90% aktiv [8, 9]. Ketolide sind unfähig, MLSB-Resistenzen zu induzieren, und sind aktiv gegenüber grampositiven Kokken wie S. aureus, S. pneumoniae und anderen Streptokokken mit induzierbarer MLSB-Resistenz. Sie besitzen zudem eine gute antibakterielle Aktivität gegenüber grampositiven Kokken, deren Resistenzmechanismus auf dem Efflux-Mechanismus beruht [4]. Die ambulant erworbene Pneumonie ist und bleibt eine der wichtigsten infektiologischen Herausforderungen an den klinisch tätigen Arzt. Die Mortalität dieser respiratorischen Infektion beträgt immer noch zwischen 5 und 15% [10]. Da zum Zeitpunkt der Diagnose der verursachende Keim praktisch nie bekannt ist, ist eine empirische Therapie unter Einschluss aller in Frage kommenden respiratorischen Erreger und mit raschem Wirkungseintritt anzustreben. Auch die American Thoracic Society und die Infectious Disease Society of America empfehlen in diesem Fall ein empirisches Vorgehen. Die häufigsten respiratorischen Keime sind S. pneumoniae, Haemophilus influenzae und Moraxella catarrhalis. Auch die Rolle der so genannten atypischen Keime wie Mycoplasma pneumoniae, Chlamydia pneumoniae oder Legionella pneumophila ist mittlerweile unbestritten. In den vergangenen 15 Jahren wurde die antibakterielle Resistenz unter den Erregern respiratorischer Infektionen zu einem weltweiten Problem. Bis in die 70er Jahre konnten Infektionen durch S. pneumoniae problemlos mit Betalaktamen behandelt werden. Seither wurde eine stetige weltweite Zunahme von Resistenzen gegenüber Penicillin G und Erythromycin beobachtet. Es besteht dringender Bedarf für antimikrobielle Substanzen, die auch gegen Penicillin G- und/oder Erythromycin A-resistente S. pneumoniae-Isolate wirksam sind. Die Klasse der Ketolide gehört zweifellos in die Gruppe neuer Antibiotika, die in der Lage sind, diese Erreger zu bekämpfen [7, 8]. Derzeit gibt es zwei Substanzen, weitere sind in der Entwicklung: In klinischer Prüfung sind das ABT 773 (Abbott) und Telithromycin (Aventis) [7]. |
| Entdeckung
der Ketolide
Im Rahmen der Bemühungen, neue Derivate ohne Potenzial für die Ausbildung einer MLSB-Resistenz zu synthetisieren, kam es zur Wiederentdeckung zweier Substanzen mit 14-gliedrigem Ring und Keto-Gruppe in Position 3: Narbomycin und Pikromycin [6, 14]. Diese beiden Substanzen können als natürliche Ketolide bezeichnet werden. Seit 1974 war bekannt, dass diese beiden Substanzen und zwei Analoga davon inaktiv, aber nicht dazu in der Lage sind, MLSB-Resistenz zu induzieren. Lange Zeit glaubte man, dass die L-Cladinose der Kernpunkt für die antimikrobielle Wirksamkeit und die Resistenz-Induzierbarkeit von Erythromycin A sei [7, 20, 23, 24, 28]. Dennoch war man der Meinung, dass die Wirksamkeit der 3-Keto-Verbindungen sehr sorgfältig neu untersucht werden sollte. So entdeckte man eine schwache, aber signifikante Wirksamkeit von Narbomycin gegen Erythromycin A-resistente, grampositive Kokken in vitro. Ausgehend von 3-Keto-6-O-Methyl-Erythromycin A, wurde eine Vielzahl von Substanzen synthetisiert, aus denen eine neue Substanzklasse semisynthetischer Derivate, die Ketolide, hervorging [11, 16]. |
| Synthese
der Ketolide
Als Ausgangspunkt wurde
ein 6-O-Methyl-Analogon von Erythromycin A verwendet. Die Cladinose
wurde in saurem Medium entfernt, und über ein 3-Hydroxy-Zwischenstadium
wurde über die modifizierte Pfitzner-Moffat-Prozedur das erste
Ketolid synthetisiert [1]. Durch diese ermutigenden Daten wurde eine weitere Erforschung der möglichen chemischen Modifikationen dieser neuen Grundsubstanz eingeleitet. So kam es zu einer Zusammenführung
der bereits zuvor entdeckten 11-, 12-Cyclocarbamat-6-Methoxy-Erythromycin
A-Derivate und der |
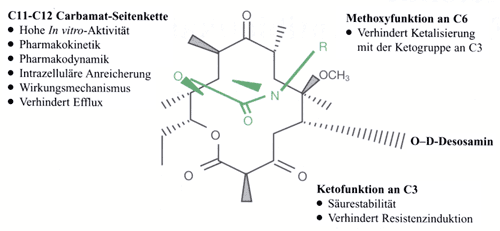
| Struktur-Wirkungsbeziehung
In Position C3 ist eine
Keto-Gruppe, in Position C6 enthält Telithromycin eine Methoxy-Gruppe
(Abb. 1).
Der Carbamat-Rest in C11 und C12 ist über eine Butyl-Seitenkette mit einem Pyridyl-Imidazol-Rest substituiert [2, 17]. Für die Wirkung sind vor allem die Ketofunktion in C3 und die Carbamat-Seitenkette in C11 und C12 verantwortlich. Die Ketofunktion in C3 vermittelt die Säurestabilität, überwindet die Makrolid-Resistenz bei Pneumokokken, verhindert die Induktion der MLSB-Resistenz und trägt zur mikrobiologischen Aktivität bei. Die Carbamat-Seitenkette bewirkt eine hohe antibakterielle Aktivität, bedingt die pharmakokinetischen und pharmakodynamischen Eigenschaften und verhindert die Induktion von Resistenzen. Zudem ist sie für die gute Gewebepenetration und die hohe intrazelluläre Anreicherung verantwortlich [12]. |
|
Wirkmechanismus Wie diese blockieren sie den Aufbau der Proteine aus Aminosäuren (Proteinbiosynthese). Der Ort der Proteinbiosynthese ist auf den Ribosomen lokalisiert. Ribosome werden im Allgemeinen
durch ihre Sedimentationskonstante in der Ultrazentrifuge charakterisiert,
die in so genannten Svedberg-Einheiten (S) 10-13s gemessen wird.
Eine Ribosomeneinheit besitzt beispielsweise eine Sedimentationskonstante
von 70S-Ribosome werden vor allem in Prokaryonten gefunden, die zytoplasmatischen Ribosome der Eukaryonten dagegen sind größer und schwerer, ihre Sedimentationskonstante liegt um 80S. Ribosome bestehen normalerweise aus einer größeren und einer kleineren Untereinheit. Die Untereinheiten der 70S-Ribosome haben im Durchschnitt Sedimentationskonstante um 50S und 30S. Diese Untereinheiten sind nur bei der Translation (Elongation) während der Proteinsynthese miteinander verbunden. Nach Freisetzung der fertigen Polypeptidkette trennen sich die Untereinheiten wieder. Aus den 50S-Untereinheiten lassen sich zwei RNS-Molekülarten isolieren, und zwar eine mit einer Sedimentationskonstante von 23S und eine kleinere von 5S. Die 30S-Untereinheit des Ribosoms enthält ein rRNS-Molekül mit einer Sedimentationskonstante von 16S [12]. Um die Proteinsynthese in Gang zu setzen, müssen sich die 30S- und die 50S-Untereinheiten der Ribosome zusammenlagern. Die Reihenfolge der Aminosäuren im Protein wird durch die Nukleotidsequenz der mRNA festgelegt und durch das Ribosom umgeschrieben (translatiert). Transfer-RNA-Moleküle (tRNA), die mit einer Aminosäure beladen sind, fügen die Aminosäuren entsprechend der Sequenz der mRNA zusammen. Der Peptidfaden wird durch einen Translationskanal durch die ribosomale RNA nach außen transportiert. In diesen Kanal lagern sich diese Antibiotika ein und verhindern so die Translokation. Jedoch gibt es genau bei dieser Einlagerung Unterschiede zwischen den einzelnen Antibiotika: Ketolide treten zwar mit denselben Stellen in Wechselwirkung wie die Makrolide, unterscheiden sich aber von den Makroliden in der Stärke der Wechselwirkung (Abb. 2).
Darüber hinaus besteht eine direkte Wirkung auf die Bildung der 50S- und 30S-Untereinheiten der Ribosome. Die Ketolide besitzen somit einen dualen Wirkmechanismus. Orte der Wechselwirkung sind das Adenin 2058 in der Domäne V der 23S rRNA und zusätzlich an Adenin 752 in der Domäne II der 23S rRNA [12]. Dies führt dazu, dass einer der entscheidenden Schritte, der für die weitere Anknüpfung von Aminosäuren an die wachsende Peptidkette (Translokation) verantwortlich ist, blockiert wird und die tRNA demzufolge zerfällt (dissoziiert). Die Peptidkette bleibt unvollständig, die Proteinsynthese bricht ab. Die Keto-Gruppe an C3 und die Carbamatgruppe an C11/C12 sorgen zusätzlich für eine mindestens zehnfach festere Bindung an die ribosomale RNA, als es für die Makrolide gemessen wurde. Im Vergleich zu Erythromycin bindet Telithromycin etwa 10-mal stärker an MLSB-sensible Ribosome und mehr als 25-mal stärker an die MLSB-resistenten Ribosome [18, 21] (Abb. 3).
|

| Welchen
Vorteil bietet dieser zweifache Wirkmechanismus?
Bei Pneumokokken, den häufigsten Erregern bei Atemwegsinfektionen in der Praxis, nimmt die MLSB-Resistenz zu. Diese Resistenz beruht darauf, dass eine Bindungsstelle am Ribosom, die Domäne V, verändert ist. Chemisch gesehen wird an die Bindungsstelle ein Methylrest angehängt. Diese Veränderung kann entweder durch den Zuckerrest, die Cladinose, an Position 3 der MLSB-Antibiotika ausgelöst werden (Induktion) oder seltener durch spontane Veränderungen (Mutation). An der veränderten Bindungsstelle können sich z. B. Makrolide nicht mehr anheften, um die Proteinsynthese zu blockieren. An der zweiten Bindungsstelle (Domäne II) am Ribosom haften MLSB-Antibiotika gar nicht bzw. nur sehr schwach, sodass über die Domäne II kein Effekt vermittelt wird. Telithromycin bindet auch an dieser zweiten Stelle mit hoher Affinität. Durch diesen zweifachen Wirkansatz wird die Proteinbiosynthese wirkungsvoll blockiert [18, 21]. Die neue Ketolid-Struktur bringt noch einen weiteren Effekt mit sich. Der Austausch des Zuckers durch die Keto-Gruppe macht eine Induktion von Resistenzen in der Zukunft unwahrscheinlich, ermB-kodierte Resistenzen werden nicht aktiviert. Außerdem wurde nachgewiesen, dass die Ketolide aufgrund ihrer Carbamat-Seitenkette die bakterielle Effluxpumpe sehr viel schwächer induzieren als z. B. Erythromycin und sich demzufolge in höherer Konzentration im Zellinneren ansammeln können. Aktuelle Untersuchungen lassen darauf schließen, dass diese günstige Resistenzsituation vergleichsweise stabil ist: So wurden Erythromycin A-sensitive Pneumokokkenstämme sequentiell mit subinhibitorischen Konzentrationen von Telithromycin behandelt. Bei diesen Stämmen konnten selbst nach mehreren Durchgängen keine Isolate mit Resistenzen gegen das Ketolid selektioniert werden [13, 15].
|
| Mikrobiologisches
Wirkungsspektrum
Ketolide zeigen eine besonders gute Wirkung gegen die häufigsten Erreger respiratorischer Infektionen. Dazu gehören S. pneumoniae, H. influenzae und Moraxella catarrhalis. Darüber hinaus wirken sie bei atypischen/intrazellulären Erregern wie Legionellen, Chlamydien und Mykoplasmen [5, 7, 29, 30]. Sie wirken sowohl gegen
Penicillin-empfindliche als auch Penicillin- und Erythromycin-resistente
Stämme von S. pneumoniae [7] (Tab. 1).
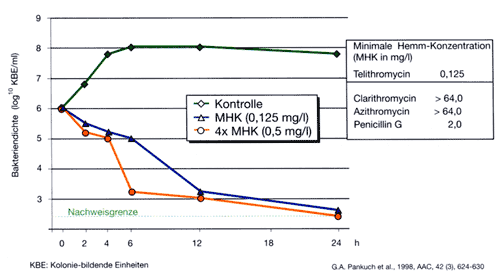
Bemerkt werden muss,
dass die Ketolide einen bakteriziden Effekt aufweisen. Weiters besteht
ein starker postantibiotischer Effekt [22, 26] (Abb. 4).
|
| Pharmakokinetik
Die pharmakokinetischen Eigenschaften der Ketolide wie Telithromycin zeichnen sich durch eine schnelle Anflutung (tmax), hohe Serumspiegel (cmax), hohe Konzentrationen über die Zeit im Plasma (AUC), lange terminale Halbwertszeit (t1/2) und hohe Gewebekonzentrationen aus [25]. Nach einmaliger Gabe von 800 mg Telithromycin werden maximale Plasmakonzentrationen von 1,99 mg/l nach 1 Stunde erreicht. Die orale Bioverfügbarkeit beträgt 57% [27]. Die Resorption wird durch gleichzeitige Nahrungsaufnahme nicht verändert. Die terminale Halbwertszeit beträgt 10,6 h. Ketolide penetrieren
schnell in die entzündlichen Gewebe und reichern sich dort
in hohen Konzentrationen an. Die Konzentrationen am Wirkort übersteigen
die Konzentrationen im Plasma [12] (Tab. 2).
|
| Zusammenfassung
Die steigenden bakteriellen Resistenzen verschiedenster Erreger gegenüber Betalaktamen und Makroliden rechtfertigen den Einsatz neuerer Substanzen, die auch gegen resistente Erreger wirksam sind. Die neue Gruppe der Ketolide und insbesondere Telithromycin eignen sich hervorragend für die empirische Therapie ambulant erworbener respiratorischer Infektionen. Das hervorragende Wirkprofil, der Mangel an Induktion von Resistenzen, die ausgezeichnete Pharmakokinetik und die gute Verträglichkeit von Telithromycin lassen diese Substanz als unverzichtbaren Bestandteil eines modernen Infektionsmanagements erscheinen. |
|
|
Anschrift
des Verfassers: |