| Dosierung von
Antibiotika im Kindesalter - Schnittstelle zwischen Pharmakokinetik
und Pharmakodynamik |
| J.P. Guggenbichler
Univ.-Klinik für Kinder und Jugendliche der Universität
Erlangen/Nürnberg
(Vorstand: Univ.-Prof. Dr. W. Rascher) |
Schlüsselwörter:
Pharmakokinetik, Pharmakodynamik,
Resorptionseinbuße, subinhibitorische Konzentrationen |
|
Zusammenfassung
Es bestehen
erhebliche Unterschiede in der Resorption, Verteilung und im
Metabolismus von Antibiotika in verschiedenen Lebensabschnitten.
Durch eine beschleunigte Peristaltik im Säuglingsalter
sowie bei verschiedenen Grundkrankheiten kommt es zu Resorptionseinbußen
von 50%, gemessen an der Fläche unter der Resorptionskurve
und der im Harn wiederentdeckten Wirkstoffmenge. Auch Resorptionseinbußen
durch gleichzeitige Verabreichung von Nahrung sind zu berücksichtigen.
Dadurch kann man nicht mit einer regulären Pharmakokinetik,
wie sie bei Erwachsenen zur Definition von effektiven Tagesdosen
und Dosierungsintervallen geführt hat, rechnen. Ein weiterer
wichtiger Faktor bei der Infektionsbehandlung von Säuglingen
und Kleinkindern besteht in der Beachtung der Pharmakodynamik
von Antibiotika, d.h. Wirkstoffkonzentrationen auch über
einen ausreichenden Zeitraum am Infektionsort aufrechtzuerhalten.
Für das Kindesalter werden Wirkstoffkonzentrationen über
dem MHK-Wert von 75-90% der Zeit bis zur nächsten Dosis
empfohlen. Dies resultiert in einer raschen Keimelimination
und klinischen Heilung sowie Verhinderung der Resistenzentwicklung.
Im Gegensatz dazu sind subinhibitorische Konzentrationen von
Antibiotika mit verzögerter klinischer Besserung, dem Übergang
in eine chronisch schwelende Infektion und der Induktion/Selektion
von resistenten Mikroorganismen verbunden. Die Beachtung der
für Säuglinge und Kleinkinder relevanten Besonderheiten
ermöglicht eine Verbesserung der Behandlungsergebnisse.
|
Key-words:
Pharmacokinetics,
pharmacodynamics, impairment of absorption, subinhibitory concentrations |
|
Summary
Substantial
differences are seen in the absorption, distribution, metabolism
and elimination of antimicrobial substances in infants and children.
Due to a faster transit time in the intestinal tract in infants
and due to acute and chronic diarrhea, frequently seen with
infections of the respiratory tract, a reduction of absorption
of 50%, determined by the area under the curve and the urinary
recovery of the antimicrobial substance is observed. Also a
significant impairment of absorption of antibiotics with concomitant
administration of food has to be taken into consideration for
various substances. Of critical importance in the selection
and evaluation of any antiinfective drug regimen is also the
conjoint consideration of the drug action i.e. the pharmacodynamics
of the antibiotic. This means that adequate concentrations of
an antimicrobial substance must be present at the site of the
infection for a sufficient period of time. For children optimum
eradication of microorganisms is achieved by surpassing the
MIC for a minimum of 75 to 90% of the time until the next dose.
This results in a rapid eradication of the infectious agent,
fast improvement of the clinical condition and the prevention
of emergence of resistant microorganisms. In contrast subinhibitory
concentrations result in functional disturbances with chronic
smolderig infections and selection of less sensitive or resistant
microorganisms. The observation of this unique situation for
infants and children has been largely neglected in the past
for the determination of the total daily dose and dosage intervals
in these age groups. Proper attention to the pharmacokinetic-pharmacodynamic
interface unique to children results in an improved clinical
outcome and prevention of therapeutic failures.
|
Einleitung
Infektionen
spielen in der Kinderheilkunde eine wesentliche Rolle. Es ist
selbstverständlich, dass die erfolgreiche Behandlung einer
Infektion die Wahl eines wirksamen Antibiotikums voraussetzt.
Sie erfolgt nach Isolierung des Keimes durch die Empfindlichkeitsprüfung
mittels Hemmhofbestimmung oder MHK-Testung. Meist erfolgt die
Wahl eines Antibiotikums jedoch nach klinischen Gesichtspunkten
noch vor dem Vorliegen mikrobiologischer Ergebnisse als empirische
oder kalkulierte Therapie. Dies ist jedoch in den letzten Jahren
durch das Auftreten resistenter Mikroorganismen auch im ambulanten
Bereich erheblich schwieriger geworden [1, 2].
Der therapeutische
Erfolg der Behandlung einer Infektion, aber auch die Entwicklung
resistenter Mikroorganismen werden entscheidend dadurch beeinflusst,
dass antimikrobiell wirksame Konzentrationen im Gewebe, in Körperhöhlen,
in Körperflüssigkeiten wie dem interstitiellen Flüssigkeitskompartment
und in der die Mukosa bedeckenden Schleimschicht (Epithelial
Lining Fluid, ELF),
d.h. am Infektionsort für eine ausreichend lange Zeit aufrechterhalten
werden. Durch das Überschreiten von Wirkstoffkonzentrationen,
die über dem MHK-Wert liegen, ist eine schnelle und effiziente
Elimination der pathogenen Mikroorganismen möglich, woraus
ein rascher Rückgang der Entzündungsreaktion resultiert.
Damit ist einerseits eine rasche klinische Heilung und die Vermeidung
des Übergangs einer akuten Infektion in eine chronisch
schwelende Infektion verbunden. Zusätzlich kann die Induktion
und Selektion resistenter Mikroorganismen verhindert werden
[3].
Eine wichtige
Orientierungsgröße für die Wirksamkeit eines
Antibiotikums ist die Pharmakokinetik, d.h.
die Bioverfügbarkeit eines Medikamentes, die durch die
Messung der Serum- und Gewebskonzentrationen und ihre Beeinflussung
durch Resorptions-, Diffusions- und Exkretionsvorgänge
bestimmt wird. Die Pharmakodynamik beschreibt
einerseits die Wirkung eines Antibiotikums auf den Keim, vor
allem aber den Zeitraum, während dem ausreichende Wirkstoffkonzentrationen
am Ort der Infektion für eine erfolgreiche Keimelimination
notwendig sind.
Dabei unterscheiden
sich alle oben genannten Parameter bei Kindern erheblich von
denen Erwachsener. Aber selbst im Kindesalter sind wesentliche
Unterschiede in den einzelnen Lebensabschnitten – z.B.
zwischen Früh- und Neugeborenen, Säuglingen, Kleinkindern
zwischen 2 und 6 Jahren, Schulkindern und Adoleszenten –
zu beobachten, bei denen sich die pharmakologischen und pharmakokinetischen
Parameter dem Erwachsenenalter annähern. Zudem sind besondere
Grundkrankheiten zu berücksichtigen, die die oben genannten
Parameter zusätzlich überlagern [4].
|
| Pharmakokinetik
/ Bioverfügbarkeit
Die Pharmakokinetik
umfasst nach der klassischen Pharmakologie die Beschreibung
der Absorption, der Verteilung (Distribution),
des Metabolismus und der Elimination
(ADME) von Medikamenten [5]. Bei der Verabreichung
von Antibiotika ist dies jedoch in einem breiteren Sinn zu sehen,
da zusätzliche „Variable“ wie der Mikroorganismus
mit einer unterschiedlichen Empfindlichkeit, Inokulumgröße,
die Entzündungsreaktion sowie die spezifische und unspezifische
körpereigene Abwehr dazukommen. Zudem besteht in den verschiedenen
Lebensabschnitten ein deutlicher Unterschied im Gesamtkörperwasser
sowie in der Zusammensetzung der einzelnen Kompartments, was
einen wesentlichen Einfluss auf die Wirkstoffkonzentration hat:
Während bei Erwachsenen das Gesamtkörperwasser 60%
und das extrazelluläre Flüssigkeitskompartment ca.
26% des Körpergewichtes ausmachen, beträgt dies bei
Frühgeborenen < 1.500 g GG 80% und 45% respektive. Bei
reifen Neugeborenen geht das extrazelluläre Flüssigkeitskompartment
auf 40% zurück und pendelt sich im Laufe des ersten Lebensjahres
auf 26-28% des Körpergewichts ein. Dementsprechend ist
das Verteilungsvolumen von ß-Laktam-Antibiotika in diesen
Lebensabschnitten um > 40% größer und es sind
die erreichbaren maximalen Wirkstoffkonzentrationen bei äquimolarer
Resorption um den entsprechenden Faktor niedriger [6].
Eine Vielzahl
von pharmakokinetischen Parametern wie die Absorption und die
Bestimmung der Invasionshalbwertszeit, die Fläche unter
der Konzentrationskurve (AUC) sowie die Bestimmung der im Harn
wiedergefundenen Wirkstoffmenge als Maß der Bioverfügbarkeit
eines Medikamentes, die Eliminationshalbwertszeit, die Verteilung
in verschiedenen Körper-Kompartmenten und das Verteilungsvolumen,
der „First Pass Effect“ wie auch die Biotransformation
von Prodrugs im Darmlumen sind für die Wahl der Tagesdosis
und der Dosierungsintervalle zu beachten.
Alle diese
Parameter bestimmen – abgesehen von unterschiedlichen
Empfindlichkeiten von Mikroorganismen – die Wirksamkeit
und Sicherheit von Antibiotika, die Wahl einer korrekten Tagesdosis
und die Dosierungsintervalle.
Orale
Therapie und Bioverfügbarkeit
Vor allem
im ambulanten Bereich erhält die Mehrzahl der Patienten
Antibiotika oral, wobei das Ausmaß von Resorption und
Bioverfügbarkeit wesentlich zum therapeutischen Erfolg
beiträgt. Untersuchungen der Bioverfügbarkeit erfolgten
bisher im Rahmen der Zulassungsuntersuchungen von Antibiotika
an gesunden, nüchternen Probanden. In der Praxis werden
die Präparate an Säuglinge und Kleinkinder mit verschiedenen
Grundkrankheiten ohne Rücksicht auf den Füllungszustand
des Magens verabreicht.
Die Resorption hängt von |
- dem
Füllungszustand und der Entleerungszeit des Magens,
- dem pH-Wert im Magen,
- der intestinalen Oberfläche und Permeabilität,
- der Peristaltik des Dünndarms,
- der Durchblutung im Splanchnikusgebiet,
- der mikrobiellen Besiedelung |
| ab
[7]. |
Bei der
Erhebung pharmakokinetischer Daten von Säuglingen und Kleinkindern
ergibt sich eine Reihe von Schwierigkeiten. Bei den wenigen
bisher in der Literatur zur Verfügung stehenden Untersuchungen
konnten erhebliche Unterschiede in den einzelnen Lebensabschnitten
beobachtet werden, was die Notwendigkeit entsprechender klinischer
und pharmakologischer Untersuchungen unterstreicht. Aus ethischen
Gründen ist die Erhebung eines kompletten pharmakokinetischen
Profils mit Bestimmung der Invasions- und Eliminationshalbwertszeit,
des Verteilungsvolumens etc. oft nicht möglich, da dies
Blutabnahmen von 2 ml in 1/2-stündlichen Intervallen bis
zu 6 Stunden erfordern würde.
Als
Ausweg aus diesem Dilemma bieten sich an:
a) Die
Untersuchung erfolgt im Rahmen einer Therapieüberwachung
als therapeutisch gerechtfertigtes Drugmonitoring. Die Konzentrationsbestimmung
erfolgt aus Einzelproben von verschiedenen Patienten, die in
bestimmten Zeitabständen entnommen werden. Einzelwerte
von zahlreichen Patienten, die zu unterschiedlichen Zeiten gewonnen
wurden, werden gepoolt und ergeben ein Gesamtbild der Bioverfügbarkeit
eines Präparates. Diese Daten sind für die Beantwortung
fast aller klinisch relevanter Fragestellungen ausreichend [8,
9].
b) Die
Reduktion des Probenvolumens und die Bestimmung der Serumkonzentration
aus einer für Kinder akzeptablen geringen Blutmenge. Bereits
mit einem Probenvolumen von 50 µl konnte mit einer mikrobiologischen
Methode in einem Doppeldiffusionsagar die Konzentrationsbestimmung
mit Mehrfachbestimmungen durchgeführt werden, wobei die
Nachweisgrenze für ß-Laktam-Antibiotika bei 0,1 µg/ml
und einer Fehlerquote <10% lag. Diese Blutmenge wurde –
gleichzeitig mit einer diagnostisch notwendigen Blutabnahme
– aus einem Fingerprick abgenommen, wobei die Stichstelle
mit Heparin Vaseline verschlossen wurde. Dies erlaubte Mehrfachentnahmen
ohne erneuten Stich. Die Bestimmung der im Harn wiederentdeckten
Wirkstoffmenge ist ohne wesentliche Belastung für den Patienten
möglich. Die Gewinnung des Harnes und die genaue Bestimmung
der Harnmenge über die entsprechenden Zeiträume bedürfen
jedoch bei Säuglingen und Kleinkindern einer sorgfältigen
Betreuung. Das Legen eines Dauerkatheters ist nicht zulässig,
aber auch nicht nötig. Diese Methode ist nur bei Präparaten
mit vorwiegend renaler Elimination bzw. bei bekannter renaler
Eliminationsmenge bei Erwachsenen oder älteren Kindern
zielführend (Abbildung 1) [10].
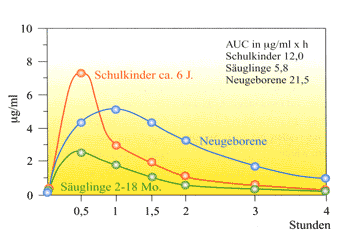
Abbildung
1: Unterschiede in der Bioverfügbarkeit
zwischen den einzelnen Lebensabschnitten bei einer Dosierung
des Kaliumsalzes von Phenoxymethylpenicillin von 12.500
IE pro Kilogramm Körpergewicht [11]
|
Schulkinder
im Alter von ca. 6 Jahren resorbieren Oralpenicillin sehr gut,
die maximale Serumkonzentration wird bereits nach 30 Minuten
erreicht, die Halbwertszeit beträgt ca. 50 Minuten. Die
Bioverfügbarkeit bei Schulkindern entspricht weitgehend
der Bioverfügbarkeit von Adoleszenten und Erwachsenen.
Neugeborene haben zwar wegen des größeren Verteilungsvolumens
niedrigere Spitzenkonzentrationen, wenn man jedoch die im Harn
ausgeschiedene Wirkstoffmenge als Maß der Bioverfügbarkeit
heranzieht, resorbieren Neugeborene sogar um ca. 40% mehr als
Schulkinder. Dies beruht möglicherweise auf einer erhöhten
Membranpermeabilität des Intestinaltraktes bei Früh-
und Neugeborenen mit besserer Penetration des Wirkstoffes im
Darm. Als weitere Möglichkeit kommen auch eine verzögerte
Magenentleerung und eine verminderte intestinale Motilität
im Neugeborenenalter in Betracht. Auch die Fläche unter
der Kurve ist bei Neugeborenen größer. Dies hängt
nicht zuletzt mit einer durch die Unreife der Nierenfunktion
bedingten verzögerten renalen Elimination zusammen. Bisweilen
ist aber die Darmmotilität unvorhersagbar, wodurch eine
exakte Vorausberechnung der Resorption nicht möglich ist
[12].
Die Bioverfügbarkeit
hängt neben der Membranpermeabilität wesentlich von
der Größe der resorptiven Oberfläche und der
Peristaltik ab. Oralpenicilline und Aminopenicilline werden
im Magen, im Duodenum und in den obersten 25-30 cm Jejunum resorbiert.
Da bei Säuglingen zwischen dem 2. und dem 15. Lebensmonat
eine um das 2- bis 3fach beschleunigte Passagezeit besteht,
kommt es zu einer substanziellen Verminderung der Resorptionsmenge
[13].
Auch bei
Azlozillin, Amoxicillin + Clavulansäure sowie bei Sulbactam
Ampicillin konnte eine ähnliche Resorptionsminderung in
diesem Lebensabschnitt festgestellt werden (Tabelle 1).
Tabelle
1: Mittelwert aus 12 Patienten, 2 Stunden nach
Gabe von jeweils 17,5 mg/kg KG des Antibiotikums
| Antibiotikum |
Alter |
| |
3
- 15 Monate |
6
Jahre |
|
| Amoxicillin
+ Clavulansäure |
2,20 µg/ml |
6,5 µg/ml |
| Sulbactam
Ampicillin |
1,36
µg/ml* |
5,0
µg/ml |
|
| *
Bei 3 von 12 Patienten < 12 Monate lag die
Wirkstoffkonzentration unter der Nachweisgrenze
von 0,20 µg/ml [14]. |
|
|
Cefalexin,
Cefaclor, Cefadroxil und Loracarbef werden im gesamten Intestinaltrakt
resorbiert. Dabei besteht keine Resorptionsminderung im Säuglingsalter.
Für oral verabreichte Cephalosporine der III. Generation
wie Cefixim, Ceftibuten, Cefpodoxim-Proxetil werden unterschiedliche
Resorptionsraten beschrieben. Nach Gabe von Cefixim und Ceftibuten
werden nur ca. 50% der verabreichten Dosis, von Cefetamet-Pivotil
und Cefpodoxim-Proxetil werden 75% der verabreichten Menge resorbiert
[15]. In der Literatur wird über eine verminderte Bioverfügbarkeit
von Prodrugs = veresterten Substanzen auf Grund der im frühen
Säuglingsalter verminderten Ausstattung mit intestinalen
Hydrolasen berichtet. Dies konnte von uns in Einzeluntersuchungen
in Form eines Drug-Monitorings bei Säuglingen zwischen
3 und 6 Monaten für Cefpodoxim-Proxetil nicht bestätigt
werden [16].
Makrolid-Antibiotika,
vor allem die neueren Makrolide wie Roxithromycin, Clarithromycin
und Josamycin, zeigen eine gute Bioverfügbarkeit in Form
von bakterizid wirksamen Serumkonzentrationen. Im Gegensatz
zu ß-Laktam-Antibiotika, die sehr rasch über die
Blutbahn in das interstitielle Kompartment verteilt werden,
füllen Makrolid-Antibiotika nach der Resorption zuerst
das interstitielle und intrazelluläre Flüssigkeitskompartment
auf, und erst der nicht gewebegebundene „Überlauf“
ist im Blut nachweisbar. Daher sind Gewebskonzentrationen um
das 100fache, Konzentrationen in der ELF um das 10fache höher
als die Serumkonzentrationen. Roxithromycin zeichnet sich durch
die höchsten Serumkonzentrationen (10 µg/ml Spitzenkonzentration),
Clarithromycin durch die höchsten ELF- (50 µg/ml)
und Gewebskonzentrationen (500 µg/g alveoläre Makrophagen)
aus [17]. Azithromycin hingegen wird sowohl als Kapsel als auch
in Saftform schlecht resorbiert und resultiert in Serumkonzentrationen
von ca 0,1-0,25 µg/ml und ELF-Konzentrationen um 1-2 µg/ml.
Die intrazellulären Wirkstoffmengen z.B. in alveolären
Makrophagen betragen 40 µg/g alv. Makrophagen und überschreiten
die MHK-Werte für Infektionen der Atemwege durch intrazelluläre
Mikroorganismen. Trimethoprim und Trimethoprim-Sulfonamid-Kombinationen
besitzen eine nahezu 100%ige Bioverfügbarkeit [18].
Resorptionsbeeinflussung durch Nahrungsmittel
Gleichzeitige
Nahrungsaufnahme – insbesondere von Milch mit einem hohen
Kalziumgehalt – führt zu substanziellen Resorptionseinbußen
von ß-Laktam-Antibiotika. Dies ist gerade im Säuglingsalter
mit den üblichen 5 Milchmahlzeiten von besonderer Relevanz.
Abbildung
2 zeigt die Resorptionseinbußen von Penicillin VK bei
Schulkindern nüchtern und nach einer Milchmahlzeit.
Abbildung
2: Resorptionseinbußen von Penicillin VK
bei Schulkindern nüchtern und nach einer Milchmahlzeit
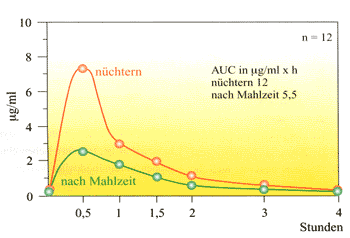
|
Penicillin
VK liegt als Salz vor, dissoziiert aber im Magen zu Phenoxymethylpenicillin
und Kalzium. Wenn gleichzeitig Milch mit dem Antibiotikum verabreicht
wird, rekombiniert sich das Phenoxymethylpenicillin mit dem
im Überschuss in der Milch vorhandenen Kalzium, und es
entwickelt sich ein schwer lösliches Kalziumsalz, das schlecht
resorbiert wird. Auch Cefaclor wird in der Bioverfügbarkeit
– individuell unterschiedlich zwischen 10% und >50%
– durch die Nahrung beeinträchtigt. Es werden jedoch
bei weitem nicht alle Antibiotika in ihrer Bioverfügbarkeit
verändert. Das Benzathinsalz des Penicillin V wird, da
weniger gut wasserlöslich, unabhängig vom Füllungszustand
des Magens resorbiert; Cefalexin und Loracarbef werden in ihrer
Bioverfügbarkeit nicht beeinflusst. Auch die Resorption
von Cefpodoxim-Proxetil wird durch gleichzeitige Verabreichung
von Nahrungsmitteln nicht beeinflusst. Makrolid-Antibiotika
zeigen mit Ausnahme von Azithromycin bei gleichzeitiger Verabreichung
mit Milch sogar höhere Wirkstoffkonzentrationen [19, 20].
Auch die
Art der Nahrung hat einen Einfluss auf die Resorption: Klare
Flüssigkeit und Fruchtsäfte steigern die Resorption
von ß-Laktam-Antibiotika, Fett und Eiweiß beeinträchtigen
die Resorption stärker als Kohlenhydrate.
Resorptionsbeeinflussung
bei verschiedenen Grundkrankheiten
Wie bereits
beschrieben, wird die Resorption oral verabreichter Antibiotika
durch verschiedene Grundkrankheiten wesentlich beeinflusst.
So hat die Durchblutung des Splanchnikusgebiets für die
Resorption einen entscheidenden Einfluss. Bei Fieber über
39,5°C wird Blut in die Peripherie zur Wärmeabstrahlung
umgeleitet und es resultiert eine Minderdurchblutung im Intestinaltrakt
mit einer Resorptionseinbuße von bis zu 40%.
Auch eine
Änderung der Peristaltik zeigt einen erheblichen Einfluss
auf die Resorption: Akute und chronische Durchfallerkrankungen,
oft Begleiterscheinungen von Infektionen der oberen und unteren
Luftwege oder von Harnwegsinfektionen, führen zu einer
erratischen Resorption, insgesamt jedoch zur deutlichen Einschränkung
der Bioverfügbarkeit, wobei die Spitzenkonzentrationen
um 50%, die Fläche unter der Kurve um bis zu 25% reduziert
ist [21].
Bei chronischen
Durchfallerkrankungen, wie z.B. bei Zottenatrophie durch Zöliakie,
beobachtet man ebenfalls eine Verminderung der Resorption um
50% insgesamt, durch Änderung der Magenentleerung können
aber noch nach Stunden unerwartet Resorptionsspitzen auftreten
[22].
Bei Mukoviszidose
wurde als Ursache für niedrigere Serumspitzenspiegel eine
Resorptionsminderung durch das zähflüssige Intestinalsekret
vermutet. Weitere Untersuchungen konnten jedoch die beobachteten
niedrigen Serumkonzentrationen auf eine beschleunigte renale
Elimination für Isoxazolylpenicilline und Aminopenicillin
zurückführen. Cephalosporine werden zwar langsamer
resorbiert und resultieren in niedrigeren Serum-Spitzenkonzentrationen,
die Fläche unter der Kurve und die im Harn wiederentdeckte
Menge entsprechen jedoch denen der Kontrollpatienten [23].
Es ist
von großer Bedeutung, bei der Wahl der Tagesdosis und
der Dosierungsintervalle diese resorptionsmindernden Faktoren
zu berücksichtigen. Bei oraler Verabreichung von Antibiotika
fehlt einerseits die nicht resorbierte Wirkstoffmenge am Infektionsort,
andererseits führt sie im Intestinaltrakt zur Störung
der normalen Flora und zu osmotischer Diarrhoe. Damit ergibt
sich ein „Circulus vitiosus“ aus verminderter Resorption,
gesteigerter Peristaltik und damit wieder verminderter Resorption.
Halbwertszeit
Durch die
im frühen Säuglingsalter bestehende Unreife der Leber
kommt es bei verschiedenen Präparaten zu einer verzögerten
Glukuronierung und Ausscheidung und damit zu einer verlängerten
biologischen Halbwertszeit. Die Unreife der Nierenfunktion bei
Früh- und Neugeborenen bzw. eine Dehydratation durch Erbrechen
im Rahmen eines Infektes oder einer begleitenden Durchfallerkrankung
führt zu einer verzögerten renalen Clearance und einer
Verlängerung der Halbwertszeit.
Präparate
mit einer unerwartet langen Halbwertszeit, wie z.B. Azithromycin
mit einer Halbwertszeit von 4 Tagen, resultieren in subinhibitorischen
Wirkstoffkonzentrationen in der ELF für ca. 5-6 Wochen.
Dies hat einen bemerkenswerten Einfluss auf die
Stabilität der normalen Rachenflora und bedingt eine Selektion/Induktion
Makrolid-resistenter Mikroorganismen (Abbildung 3) [24].
Abbildung
3: Subinhibitorische Wirkstoffkonzentrationen
führen im selektiven Fenster einerseits zu einer
verminderten Virulenz und einer besseren Phagozytierbarkeit
von Mikroorganismen, andererseits zu einer erheblichen
Steigerung der Resistenz
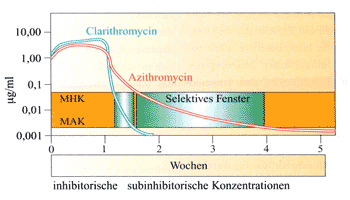
|
Metabolismus
Die Metabolisierung
eines Medikamentes ist vielfach notwendig, damit ein Präparat
renal oder über die Galle besser ausgeschieden werden kann.
Dies bedeutet eine Veränderung des Moleküls, meist
eine Steigerung der Hydrophilie. Durch Veränderungen des
Moleküls, z.B. Phosphorylierung von Acyclovir, kommt es
zu einer Aktivierung des initial inerten Wirkstoffes [25]. Bisweilen
bleibt die antimikrobielle Wirksamkeit auch bei Metaboliten
(Josamycin und Clarithromycin) oft mit synergistischer Wirksamkeit
erhalten. Meist kommt es jedoch zur Abnahme – Cefotaxim
wird zum weniger aktiven Desacetylcefotaxim – oder zur
Aufhebung der Wirksamkeit [26].
Wenig ist
über klinische Konsequenzen aus einem veränderten
Metabolismus antimikrobieller Substanzen im frühen Säuglings-
und Kleinkindesalter im Vergleich zu älteren Kindern bekannt.
Bei Neugeborenen ist eine verminderte Eiweißbindung von
antimikrobiellen Substanzen beschrieben, die zu Verdrängungsmechanismen
bei Bilirubin führt. Bei intravenöser Verabreichung
von Ceftriaxon mit einer Eiweißbindung von 98% kann dies
möglicherweise zu einem Kernikterus beitragen, bei oral
verabreichten Präparaten spielt dies jedoch nur eine untergeordnete
Rolle. Viele Medikamente, einschließlich Antibiotika,
werden durch Cytochrom P-450 metabolisiert. Da dieses Enzym
induzierbar ist, bestehen neben einer großen intraindividuellen
Variabilität auch erhebliche Unterschiede in den einzelnen
Lebensabschnitten und Wechselwirkungen mit anderen Medikamenten
[27]. Die verminderte Glukuronierung von oral verabreichten
Präparaten in der Leber im Neugeborenenalter ist klinisch
nicht relevant. Zu beachten ist eine höhere Toxizität
von Trimethoprim im ersten Trimenon, was die Verabreichung dieses
Präparates in diesem Lebensalter ausschließt. Bei
Chloramphenicol wurde als schweres toxisches Erscheinungsbild
das Gray-Syndrom beschrieben. Da das Präparat gegenwärtig
nur noch bei besonderen Indikationen verabreicht wird, spielt
diese Nebenwirkung heute praktisch keine Rolle mehr.
Zu beachten
ist außerdem die Interaktion von Antibiotika mit gleichzeitig
verabreichten anderen Präparaten. Die Verabreichung von
Erythromycin z.B. gleichzeitig mit Theophyllin kann zu toxischen
Theophyllinkonzentrationen mit Erbrechen und Krampfanfällen
führen. Für neuere Makrolid-Antibiotika wie Clarithromycin
oder Roxithromycin gilt diese Interaktion jedoch in einem erheblich
geringeren Maße und ist bei der üblichen niedrigeren
Dosierung von 10 mg/kg KG nicht mehr zu erwarten. Verschiedene
Makrolid-Antibiotika reduzieren jedoch die Wirksamkeit von Antiepileptika
[28, 29].
Die Annahme,
dass veresterte Präparate wie Cefuroxim-Axetil oder Cefpodoxim-Proxetil
– sog. Prodrugs – wegen der verminderten intestinalen
Enzymausstattung im ersten Trimenon weniger gut resorbiert werden,
hat sich in eigenen Untersuchungen im Rahmen eines therapeutischen
Drug-Monitorings nicht bestätigt.
Die Unreife
der Niere im Neugeborenenalter bis ins erste Trimenon hinein
in Verbindung mit einem Flüssigkeitsverlust kann zu einer
verzögerten renalen Clearance und dadurch zu einer Verlängerung
der Halbwertszeit führen.
Gewebsverteilung
Nach der
Resorption eines Antibiotikums aus dem Intestinaltrakt verteilt
sich das Antibiotikum in verschiedenen Körper-Kompartmenten.
Es ist von Bedeutung, dass ausreichend hohe aktive Wirkstoffkonzentrationen
am Infektionsort erreicht werden. Dabei ist jedoch entscheidend,
in
welchem Kompartment sich die Infektionserreger befinden. Für
intrazellulär gelegene Mikroorganismen wie Mykoplasmen
und Chlamydien, Legionellen, aber auch Listerien, Borrelien,
Leptospiren und Rickettsien, sind intrazelluläre Wirkstoffkonzentrationen
von entscheidender Bedeutung. Zur Eradikation von Mikroorganismen,
die im interstitiellen Flüssigkeitskompartment oder in
Körperhöhlen zu Infektionen führen (Staphylokokken,
Pneumokokken, H. influenzae, Streptokokken), sind besonders
interstitielle Wirkstoffkonzentrationen wichtig. Hohe, überwiegend
intrazelluläre Wirkstoffkonzentrationen sind dabei sogar
hinderlich. Zur Eradikation von Mikroorganismen an Epitheloberflächen
muss die Wirkstoffkonzentration im Epithelial-Lining-Fluid ausreichend
groß sein.
Bei der
Bestimmung von Gewebskonzentrationen muss man berücksichtigen,
dass in einer Gewebsprobe die gesamte Wirkstoffkonzentration
gemessen wird. Sie besteht aus 5% Wirkstoff im Blutkompartment,
35% im interstitiellen Flüssigkeitskompartment und 60%
im intrazellulären Kompartment. Konzentrationen im interstitiellen
Flüssigkeitskompartment werden am besten in der Hautblasenflüssigkeit
gemessen. Die verschiedenen Antibiotikagruppen verteilen sich
jedoch nicht gleichmäßig im Gewebe. Lipid-unlösliche
Substanzen, wie alle ß-Laktam-Antibiotika, verteilen sich
weitgehend im interstitiellen Flüssigkeitskompartment,
in Körperhöhlen und Exsudaten. Lipidlösliche
Substanzen wie Makrolide, Chinolone und Rifampin erreichen hohe
Wirkstoffkonzentrationen intrazellulär. Außer der
Lipid-Löslichkeit wird die Gewebsgängigkeit eines
Präparates auch von der Eiweißbindung, von aktiven
Transportmechanismen und der unterschiedlichen Fensterung des
Kapillarbettes in bestimmten Organen gesteuert. Während
ß-Laktam-Antibiotika gut ins Mittelohrsekret penetrieren
– es werden ca 30% der gleichzeitig gemessenen Serumkonzentrationen
auch im Mittelohrsekret gefunden –, besteht für die
Meningen im Rahmen der Blut-Liquor-Schranke eine besonders geringe
Durchlässigkeit. Die sog. „tight junctions“
sind jedoch bei akuten Entzündungsprozessen weit geöffnet
und erlauben eine Penetration von Antibiotika in den Liquorraum
im akuten Stadium (erster Behandlungstag) einer Meningitis von
40% gleichzeitig gemessener Serumkonzentrationen. Chloramphenicol
ist das einzige Antibiotikum, das auch zu Wirkstoffkonzentrationen
im Gehirn führt.
Granulozyten
und alveoläre Makrophagen tragen aktiv Wirkstoffe in den
Infektionsort. Dies ist jedoch mengenmäßig eher zu
vernachlässigen.
Wesentlich bei der Betrachtung der Verteilung und Gewebspenetration
von Antibiotika ist auch eine „balanzierte“ Kinetik,
d.h. eine ausreichende Wirkstoffkonzentration in allen Kompartmenten.
|
| Pharmakodynamik
Es ist bemerkenswert,
dass man gerade in der pädiatrischen klinischen Praxis
Situationen beobachtet, bei denen ein in der traditionellen
Wirksamkeitsprüfung als empfindlich getesteter Mikroorganismus
nicht eliminiert werden kann oder bei gleicher antimikrobieller
Wirksamkeit von 2 Präparaten ein deutlich besseres Behandlungsergebnis
mit dem einen Antibiotikum im Vergleich zu einem zweiten Präparat
zu beobachten ist, ohne dass sich dieses durch die traditionelle
antimikrobielle Wirksamkeitsprüfung mittels MHK-Wert erklären
ließe [30]. Die Bestimmung der minimalen Hemmkonzentration
(MHK) gibt einen wesentlichen Anhaltspunkt für die Wirksamkeit
eines Antibiotikums. Von großem Interesse ist jedoch auch,
für welchen Zeitraum Wirkstoffkonzentrationen über
dem MHK-Wert am Infektionsort aufrechterhalten werden müssen.
Dies wird als Pharmakodynamik bezeichnet und besteht in einer
Kompilation von Pharmakokinetik und MHK-Wert. Sie erfasst die
Wirkstoffmenge, die über eine bestimmte Zeitspanne am Infektionsort
nötig ist, um eine effiziente Eradikation der Bakterien
zu gewährleisten. Diese Daten sind zwar auch für die
Auswahl eines Antibiotikums, in einem viel höheren Maße
jedoch für die Wahl der Tagesdosis und der Dosierungsintervalle
von Bedeutung [31, 32].
Unterscheidung in zeitabhängige und konzentrationsabhängige
Wirksamkeit von Präparaten
Bezüglich
ihrer Wirksamkeit unterscheidet man zwischen Präparaten,
deren Wirksamkeit durch die Fläche unter der Serum- oder
Gewebskonzentrationskurve (zeitabhängige Wirksamkeit, T
>MIC), und anderen Präparaten, deren Wirksamkeit von
ihrer Spitzenkonzentration (konzentrationsabhängige Wirksamkeit)
bestimmt wird [33]. Bei zeitabhängiger Wirksamkeit wird
eine maximale Absterbegeschwindigkeit bereits bei Konzentrationen
wenig über dem MHK-Wert beobachtet, die jedoch durch eine
höhere Dosis nicht mehr steigerbar ist [34]. Als wesentliches
Merkmal ergibt sich, dass diese Konzentrationen für eine
ausreichende Zeit über dem MHK-Wert aufrechterhalten werden
müssen. Je kürzer die Zeitspanne über dem MHK-Wert
liegt, umso eher kommt es zum Wiederanwachsen der Mikroorganismen
[35]. Inwieweit ein postantibiotischer Effekt eine Rolle spielt,
ist für die Klinik noch nicht abschließend geklärt.
Es ist jedoch nicht auszuschließen, dass der postantibiotische
Effekt nicht zuletzt der Aktivität der körpereigenen
Abwehr zuzuschreiben ist.
Typische
Beispiele von Präparaten, bei denen die Fläche unter
der Kurve ausschlaggebend ist, sind ß-Laktam-Antibiotika
und Glykopeptide.
Bei den
konzentrationsabhängigen Präparaten (Cmax/MIC),
die vor allem von Aminoglykosiden und Fluorochinolonen repräsentiert
werden, ist die Dosissteigerung direkt proportional mit einer
schnelleren Keimelimination. Auch Metronidazol gehört zu
dieser Gruppe von Antibiotika. Während man bei zeitabhängigen
Präparaten durch eine Verkürzung der Dosierungsintervalle
bis hin zur Dauerinfusion eine Verbesserung der antimikrobiellen
Wirksamkeit erzielt, ist bei den konzentrationsabhängigen
Präparaten durch eine Steigerung der Initialdosis vor allem
zu Beginn der Behandlung eine bessere Wirksamkeit zu erzielen.
Dadurch ist eine Verlängerung der Dosierungsintervalle
möglich [36]. Bei subinhibitorischen Konzentrationen dieser
Präparate kommt es jedoch zu einer Steigerung der adaptiven
Resistenz.
Makrolid-Antibiotika
liegen zwischen zeit- und konzentrationsabhängigen Präparaten.
Es ist einerseits eine Wirkstoffkonzentration über dem
MHK-Wert über einen ähnlichen Zeitraum wie bei der
Verabreichung von ß-Laktam-Antibiotika aufrechtzuerhalten,
anderseits kann man durch Dosissteigerung eine raschere Keimeradikation
bzw. eine bakterizide Wirkung erzielen.
In
vitro-Untersuchung der Absterbegeschwindigkeit unter dem
Einfluss von in vivo beobachteten Wirkstoffkonzentrationen
In einem
noch an der Kinderklinik Innsbruck entwickelten kinetischen
Modell wurden In vitro-Wirkstoffkonzentrationen, die
in vivo erreicht werden, mit einer künstlichen
Niere nachgeahmt und stündlich Keimzahlbestimmungen durchgeführt.
Je nach Durchflussgeschwindigkeit in den Kompartmenten konnte
man Konzentrationsverläufe nachahmen, die in vivo
z.B. im Serum, im interstitiellen Flüssigkeitskompartment
oder im Mittelohrsekret beobachtet wurden. Die Keimelimination
wurde unter dem Einfluss zahlreicher Antibiotika aus verschiedenen
Wirkstoffgruppen unter dem Einfluss verschiedener Konzentrationsverläufe
bestimmt [38, 39].
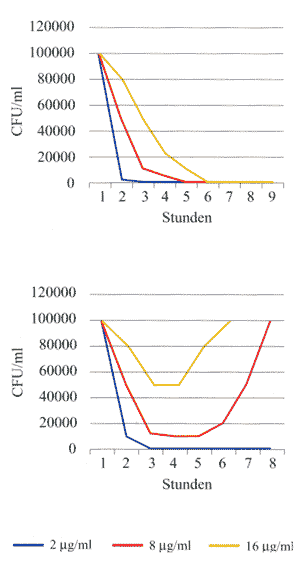
Abbildung
4a zeigt die stündliche Messung der Keimzahlen (CFU) von
Staphylococcus aureus (MHK-Wert 0,25 µg/ml) unter
2,5 µg/ml, 25 µg/ml, 250 µg/ml Cefamandol.
Die obere Kurve zeigt die Absterbegeschwindigkeit unter fixen
Konzentrationen, die untere Kurve die Absterbegeschwindigkeit
und das Wiederanwachsverhalten bei einer Eliminations-Halbwertszeit
von 1 Stunde. Es zeigt sich, dass auch bei 10-, 100- und 1.000facher
Überschreitung der MHK-Werte gleich schnelle Absterbegeschwindigkeiten
von S. aureus zu beobachten sind. Sobald der MHK-Wert
im Kulturmedium jedoch unterschritten wird, kommt es zum Wiederanwachsen
der Keime, bei niedrigeren Konzentrationen früher als bei
höheren Konzentrationen.
Abbildung
4b zeigt die Absterbegeschwindigkeit von S. aureus
unter dem Einfluss verschiedener Konzentrationen von Gentamicin.
Bei fixen Konzentrationen ist durch eine Verdoppelung der Wirkstoffkonzentration
eine erheblich beschleunigte Absterbegeschwindigkeit zu beobachten.
Bei abnehmenden Konzentrationen kann man ein Wiederanwachsen
nach Unterschreiten des MHK-Wertes beobachten. Bei einer 10
x höheren Dosis ist jedoch eine komplette Keimelimination
zu sehen.
| Abbildung
4a:
Stündliche Messung der Keimzahlen (CFU)
von Staphylococcus aureus /MHK-Wert 0,25 µg/ml)
unter 2,5 µg/ml, 25 µg/ml, 250 µg/ml
Cefamandol
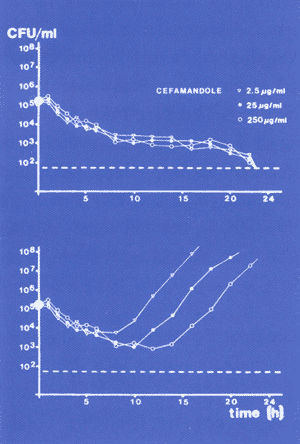
|
Abbildung
4b:
Absterbegeschwindigkeit von S. aureus unter fixen
Konzentrationen von Gentamicin (oben) und fallenden Konzentrationen
(unten), Halbwertszeit 1 Stunde
|
Gleichzeitig
wurde auch eine Beobachtung der Veränderung der Morphologie
eines Mikroorganismus unter dem Einfluss von Antibiotika durchgeführt.
Ein Antibiotikum
wird der Nährlösung (Isosensitest Bouillon + Faktor
X + NADH + Supplement B) mit 10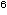 CFU/ml H. influenzae (MHK-Wert 0,25 µg/ml) zugegeben.
Im vorliegenden Experiment wurde initial eine Konzentration
von 25 µg/ml Amoxicillin gewählt, das mit einer Halbwertszeit
von 1 Stunde mittels einer künstlichen Niere eluiert wurde.
Stündlich wurden Wirkstoffkonzentrationen von Amoxicillin,
die Keimzahlen und die morphologischen Veränderungen bestimmt
(Abbildung 5).
CFU/ml H. influenzae (MHK-Wert 0,25 µg/ml) zugegeben.
Im vorliegenden Experiment wurde initial eine Konzentration
von 25 µg/ml Amoxicillin gewählt, das mit einer Halbwertszeit
von 1 Stunde mittels einer künstlichen Niere eluiert wurde.
Stündlich wurden Wirkstoffkonzentrationen von Amoxicillin,
die Keimzahlen und die morphologischen Veränderungen bestimmt
(Abbildung 5).
| Abbildung
5: Absterbekinetik von H. influenzae
unter fluktuierenden Konzentrationen von Amoxicillin in
einem kinetischen Modell
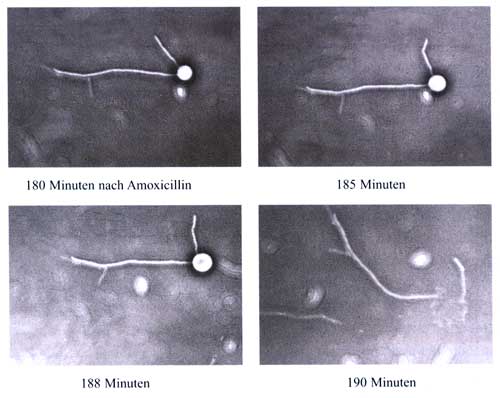
|
Die Betrachtung
der Mikroorganismen im Phasenkontrastmikroskop ergab, dass der
erste Effekt eines ß-Laktam-Antibiotikums in einer Hemmung
der Zellteilung an der vorgesehenen Teilungsstelle besteht.
Somit bildet sich ein sog. Filament, das die vielfache Länge
des ursprünglichen Mikroorganismus aufweist (Abbildung
5a).
Die nächste
Wirkung des ß-Laktam-Antibiotikums besteht im Eingriff
in den bakteriellen Stoffwechsel: Es kommt zur Blockade der
Transpeptidase, welche die festen Zellwandbausteine aus Muraminsäure
verschweißt. Dadurch entstehen Schwachstellen in der Zellwand,
und bei fortgesetztem Bakterienwachstum entstehen in dieser
Risse. Die Bakterienzelle wird an dieser Stelle nur noch durch
die elastische Zellmembran zusammengehalten. Die Ausbuchtungen
der Zelloberfläche werden als Sphäroplast bezeichnet.
In der Folge platzt auch die Zellmembran (Abbildung 5d), was
den Tod des Keimes zur Folge hat. Wenn die antimikrobielle Wirkstoffkonzentration
schon frühzeitig einen kritisch niedrigen Spiegel –
weitgehend identisch mit dem MHK-Wert – unterschreitet,
kommt es zur Aufhebung der ersten Wirkung eines Antibiotikums,
der Hemmung der Zellteilung und dadurch zur Abtrennung der Enden
eines Bakteriums.
Die aus
dem absterbenden Keim unter dem Einfluss von Antibiotika entstehenden
neuen Tochterkeime zeichnen sich durch eine geringere Menge
von Lipopolysacchariden und Lipooligosacchariden in der Zellwand
aus. Dadurch wird das Bakterium etwas weniger virulent, da diese
Zellwandbestandteile für die Entzündungsreaktion verantwortlich
sind. Gleichzeitig haben Untersuchungen ergeben, dass diese
Mikroorganismen wegen einer geringeren Elektronegativität
der Oberfläche leichter durch Phagozyten aufgenommen werden
können [40]. Ausreichende Wirkstoffkonzentrationen am Ort
der Infektion über einen entsprechenden Zeitraum können
dieses Phänomen verhindern.
Für
welchen Zeitraum der MHK-Wert für eine effiziente Keimelimination
überschritten werden muss, hängt von der erreichbaren
Wirkstoffkonzentration am Infektionsort, vom MHK-Wert des Mikroorganismus
und von der Funktionstüchtigkeit der körpereigenen
Abwehr ab. Über den notwendigen Zeitraum gibt es aber nur
Spekulationen und einzelne klinische Studien bzw. Untersuchungen
im Tiermodell, die einen Hinweis erlauben. Daten aus der Literatur
weisen darauf hin, dass der MHK-Wert am Infektionsort für
ß-Laktam-Antibiotika für mindestens 50% der Zeit
bis zur nächsten Dosis aufrechterhalten werden muss [41,
42]. Dies stimmt vor allem für erwachsene Patienten mit
intakter körpereigener Abwehr, die imstande sind, einen
durch das Antibiotikum geschädigten Mikroorganismus zu
eliminieren. Bei eingeschränkter spezifischer, aber auch
unspezifischer körpereigener Abwehr sind für eine
effiziente Keimelimination aber erheblich längere Wirkstoffkonzentrationen
>MHK-Wert am Infektionsort aufrechtzuhalten.
Wir können
heute annehmen, dass bei intakter körpereigener Abwehr
bei einem Erwachsenen oder älteren Kindern und Adoleszenten
mit einer „community acquired infection“ Wirkstoffkonzentrationen,
die den MHK-Wert für 50-60% bis zur nächsten Dosis
überschreiten, genügen. Bei Patienten, bei denen man
sich auf eine intakte körpereigene Abwehr nicht verlassen
kann, z.B. bei neutropenischen Patienten, Patienten unter Zytostatikatherapie
und bei Früh- und Neugeborenen, ist die Überschreitung
des MHK-Wertes für die gesamte Zeit bis zur nächsten
Dosis erforderlich. Bei verminderter körpereigener Abwehr
oder schweren Infektionen mit einem hohen Inokulum, die im Säuglings-
und Kleinkindesalter häufig vorliegen, ist ebenfalls eine
längere Dauer als bei Schulkindern und Adoleszenen, d.h.
ein Zeitraum von 75%-90% Überschreitung des MHK-Wertes
bis zur nächsten Dosis erforderlich.
Klinische
Beobachtung
Zwangsläufig
ergibt sich die Frage, ob eine effektive Keimelimination in
vitro mit besseren klinischen Ergebnissen korreliert. So
ist von Interesse, ob bei längerer Aufrechterhaltung bakterizider
Wirkstoffmengen in der Paukenhöhle, was mit einer rascheren
Keimelimination einhergeht, bei einem geringeren Prozentsatz
von Patienten mit einem persistierenden Tuben-Paukenhöhlenkatarrh
zu rechnen ist [43].
Ein persistierender
Tuben-Paukenhöhlenkatarrh ist ein multifaktorielles Geschehen:
Obstruktion der Tubenmündung durch vergrößerte
Adenoide, eine Tubenfunktionsstörung bei anatomischen Fehlbildungen
(Gaumenspalte, abnormer Ansatz des M. tensor veli palatini,
Knorpelschwäche) und einer spezifischen und unspezifischen
Abwehrschwäche einschließlich einer verminderten
Mukosa-Immunität spielen eine wichtige Rolle. Noch größere
Bedeutung kommt aber einer ungenügenden antimikrobiellen
Wirksamkeit sowie einer zu langsamen Eradikation von Mikroorganismen
zu [44].
Die Untersuchungen
von Gehanno bei akuter Otitis media geben dafür einen klinisch
relevanten Hinweis: Bemerkenswert an dieser Studie war, dass
alle Mikroorganismen sowohl auf Amoxicillin/Clavulansäure
als auch auf Cefpodoxim empfindlich waren und der unmittelbare
Behandlungserfolg am Therapieende mit 96% bei beiden Behandlungsregimen
gleich gut war. Trotzdem belegt diese Studie nach Gabe von Cefpodoxim
Proxetil einen erheblich niedrigeren Prozentsatz an Patienten
mit persistierendem Tuben-Paukenhöhlenkatarrh, was auch
mit einem niedrigeren Prozentsatz an Rezidiven korreliert [45]
(Tabelle 2).
| Tabelle
2 : Cefpodoxim bei akuter Otitis media, Vergleich
mit Amoxicillin und Clavulansäure, Studie 2 (6) [Gehanno
et al. 1992]
| Befunde* |
Cefpodoxim |
Amoxicillin
u.
Clavulansäure |
Unterschied |
|
Normaler
oto-
skopischer Befund |
81,1
% (90/111) |
63,8
% (60/94) |
signifikant |
|
Normales
Tympanogramm |
78,0
% (90/111) |
61,4
% (60/94) |
signifikant |
|
| Seröse
Otitis |
14,4
% (90/111) |
28,7
% (60/94) |
signifikant |
|
| *
Befunde bei Nachuntersuchung (10-20 Tage nach
Behandlungsende) |
|
|
Diese Ergebnisse
scheinen im Moment nach bisherigen Gesichtspunkten nicht erklärbar.
Vergleicht man jedoch die Interaktion Pharmakokinetik und Pharmakodynamik
bei Amoxicillin und Cefpodoxim Proxetil, kann man feststellen,
dass nach Gabe von 50 mg/kg KG Amoxicillin, aufgeteilt auf 3
Tagesdosen, der MHK-Wert für H. influenzae nur
für 50% der Zeit bis zur nächsten Dosis im Mittelohrsekret
überschritten wird, vorausgesetzt, dass das Präparat
in 8-stündlichen Intervallen verabreicht wird. Nach Gabe
von Cefpodoxim Proxetil in einer Tagesdosis von 12-15 mg/kg
KG, aufgeteilt auf 2 Dosen, beobachtet man über dem MHK-Wert
liegende Wirkstoffkonzentrationen für Moraxella catarrhalis
für 90% der Zeit bis zur 2. Dosis, bei H. influenzae,
Streptococcus pyogenes und Pneumokokken für 100%
der Zeit [46].
Abbildung
6 zeigt den Konzentrationsverlauf von Amoxicillin im Mittelohrsekret
nach Gabe von 20 mg/kg KG 8-stündlich (schwarze Linie)
und mit dem sog. „Zweitschlag“, wobei eine zusätzliche
Dosis nach 4 Stunden verabreicht wird (rote Linie). Bei einer
8-stündlichen Verabreichung wird der MHK-Wert für
H. influenzae nur für 50% der Zeit überschritten,
beim „Zweitschlag“ über 100% der Zeit in den
ersten 24 bzw. 32 Stunden.
Dieses
Phänomen wurde in einer randomisierten, prospektiven klinischen
Studie an 100 Patienten untersucht: Nach Gabe von 60 mg/kg KG
Amoxicillin, auf 2 bzw. 3 Tagesdosen aufgeteilt, wurde bei 25%
und 26,5% der Patienten resp. 18-24 Tage nach Therapieende ein
persistierender Paukenhöhlenkatarrh beobachtet. Im Gegensatz
dazu persistierte beim sog. „Zweitschlag“ mit Verabreichung
einer zusätzlichen Dosis von 20 mg/kg KG 4 Stunden nach
der ersten Gabe (80 mg/kg KG Tagesdosis am ersten Tag) nur bei
8% der Patienten ein Tuben-Paukenhöhlenkatarrh. Eine Verdoppelung
der Erstdosis kann hingegen zu einer Überschreitung des
Resorptionsmaximums führen und hat keine wesentlich höheren
Wirkstoffkonzentrationen in Serum und Paukenhöhlensekret
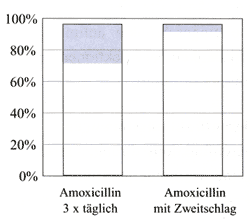
zur Folge (Abbildung 7) [47].
| Abbildung
6: Konzentrationsverlauf von Amoxicillin im Mittelohrsekret
nach Gabe von 20 mg/kg KG 8-stündlich (schwarze Linie)
und mit dem sog. Zweitschlag, wobei eine zusätzliche
Dosis nach 4 Stunden verabreicht wird (rote Linie)
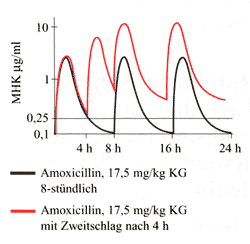
|
Abbildung
7: Behandlungsergebnisse bei akuter Otitis media:
Paukenhöhlenkatarrh 3 Wochen nach Therapieende, a)
3x tägliche Gabe, b) "Zweitschlag" 4x20
mg/kg KG
|
Bei Verabreichung
von Makrolid-Antibiotika, z.B. von Clarithromycin, wird diese
Dosierungsstrategie bereits durch die Gabe von Klacid pro®
(Verdoppelung der Dosis am ersten Tag) erfolgreich praktiziert.
Allerdings besteht für Clarithromycin kein Transportmaximum
im therapeutischen Dosierungsbereich.
|
| Rationelle
Wahl der Tagesdosis und Dosierungsintervalle unter besonderen
klinischen Bedingungen
Leberfunktionsstörungen
Eine antimikrobielle
Therapie kann einerseits eine bestehende Leberfunktionsstörung
verstärken, andererseits durch die verminderte Glukuronierung
und Ausscheidung der Substanz bei bestehender Leberfunktionsstörung
zu Kumulation führen. Nachdem bisher keine Parameter beschrieben
wurden, anhand derer eine Dosisanpassung bei eingeschränkter
Leberfunktion möglich wäre, sind Antibiotika und Chemotherapeutika,
die in der Leber metabolisiert werden, zu vermeiden. Zu diesen
Substanzen gehören in erster Linie Makrolid-Antibiotika
und Lincosamine, Rifampicin und praktisch alle Tuberkulostatika,
Tetrazykline und Metronidazol. Bei Leberfunktionsstörungen
sind meist auch die Wirkstoffkonzentrationen in der Galle vermindert,
woraus eine eingeschränkte antimikrobielle Wirksamkeit
bei Infektionen der Gallenwege resultiert.
Nierenfunktionsstörungen
Nierenfunktionsstörungen
resultieren in einer verminderten Elimination der Wirksubstanz.
Dadurch kommt es zu Kumulation und Toxizität, die wiederum
die Nierenfunktionsstörung verstärkt. So kann es bei
eingeschränkter Nierenfunktion bereits durch therapeutische
Dosen von Aminoglykosiden zu Innenohrschwerhörigkeit und
neurologischen Reaktionen mit muskulärer Hypotonie, bei
Verabreichung von Imipenem-Cilastatin zu Krampfanfällen,
und nach Gabe von Penicillin zu neuromuskulärer Hyperexzitabilität
kommen. Eine Nierenfunktionsstörung steigert die Nephrotoxizität
von Amingoglykosiden und Glykopeptiden. Die toxische Wirkung
von Aminoglykosiden kann durch Interaktion mit anderen Medikamenten,
z.B. Furosemid, durch Kompetition in der Elimination verstärkt
werden. Die gleichzeitige Gabe von Fosfomycin reduziert die
Nephrotoxizität dieser Präparate. Eine Dosisanpassung
erfolgt bei den meisten Medikamenten anhand der Kreatinin-Clearance
durch Anpassung der Tagesdosis. Die Dosisanpassung kann aber
auch dadurch erfolgen, dass als erste Dosis die Regeldosis verabreicht
wird und entsprechend der Kreatinin-Clearance die Dosierungsintervalle
verlängert werden. Die Feinabstimmung erfolgt durch die
Bestimmung der Talspiegel. Aminoglykoside, Glykopeptide und
Cephalosporine der I. Generation bedürfen im Fall einer
Nierenfunktionsstörung unbedingt einer Dosisanpassung.
|
| Schlussfolgerung
Es bestehen erhebliche
Unterschiede in der Resorption, Verteilung und im Metabolismus
von Antibiotika in verschiedenen Lebensabschnitten. Dadurch
kann man nicht mit einer regulären Pharmakodynamik, wie
sie bei Erwachsenen zur Definition von effektiven Tagesdosen
und Dosierungsintervallen geführt hat, rechnen. Dies führt
nicht selten zu subinhibitorischen Konzentrationen von Antibiotika
mit verzögerter klinischer Besserung, Übergang in
eine chronisch schwelende Infektion und Induktion/Selektion
von resistenten Mikroorganismen. Die Beachtung der für
Säuglinge und Kleinkinder relevanten Änderungen ermöglicht
eine Verbesserung der Behandlungsergebnisse.
|
Literatur:
| 1.
Baquero F.: „Trends in antibiotic resistance of respiratory
pathogens: an analysis and commentary on a collaborative
surveillance study.“ J. Antimic. Chemoth. 38 Suppl.
A (1996) 117-132. |
| 2.
Felmingham D., Grüneberg R.N.: „The Alexander
Project 1996 – 1997. Latest susceptibility data from
this institutional study of bacterial pathogens from community
acquired lower respiratory tract infections.“ J. Antimic.
Chemoth. 46 (2000) 767-773. |
| 3.
Dagan R., Klugman K.P., Craig W.A., Baquero F.: „Evidence
to support the rationale that bacterial eradication in respiratory
tract infections is an important aim of anti bacterial therapy.“
J. Antimic. Chemoth. 47 (2001) 129-140. |
| 4.
Gilman J.T., Gal P.: „Pharmacokinetic and pharmacodynamic
data collection in children and neonates. A quiet frontier.“
Clin. Pharmacokinet. 23 (1992) 1-9. |
| 5.
Abdel-Rahman S., Kearns G.L.: „The pharmacokinetic
– pharmacodynamic interface: Determinants of Anti-infective
Drug Action and Efficacy in Pediatrics.“ In Feigin,
Cherry, Demmler, Kaplan: Textbook of Pediatric Infectious
Diseases II (2004) 2965-2987. Saunders. |
| 6.
Bourne Ed: „A first Course of Pharmacokinetics and
Biopharmaceutics.“ http://gaps.cpb.ouhsc.edu 2001. |
| 7.
Ritschel W.A., Kearns G.L.: „Pediatric Pharmacokinetics.“
In Ritschel und Kearns: Handbook of Basic Pharmacokinetics.
5th Ed. American Pharmaceutical Association (1999) 304-321. |
| 8.
Reed M.D., Blumer J.L.: „Therapeutic drug monitoring
in the pediatric intensive care unit.“ Ped. Clin.
North. Am. 42 (1994) 1127-1143. |
| 9.
Kearns G.L.: „Impact of developmental pharmacology
on pediatric study design: Overcoming the challenges.“
J. Allergy Clin. Immunol. 106 Suppl. (2000) 128-138. |
| 10.
Guggenbichler J.P., Kienel G.: „Pharmakokinetische
Untersuchung mit neuen galenischen Zubereitungen von Cephalexin.“
Päd. Pädol. 13 (1978) 315-320. |
| 11.
Guggenbichler J.P.: „Resorption oral verabreichter
Antibiotika und Chemotherapeutika bei Kindern und ihre Beeinflussung.“
Päd. Pädol. 17 (1982) 565-584. |
| 12.
Leeder J.S., Kearns G.L.: „Pharmacogenetics in pediatrics.“
Ped. Clin. North. Am. 44 (1997) 55-77. |
| 13.
Weingärtner L.: „Experience with Amoxicillin
in neonates and premature babies.“ Int. J. Clin. Pharm.
Ter. Tox. 15 (1977) 184-188. |
| 14.
Renner Ch., Überall M., Guggenbichler J.P: „Unpublizierte
Beobachtungen“ (1992). |
| 15.
Hughes G.S.: „The effect of gastric pH and food on
the pharmacokinetics of a new oral cephalosporin, cefpodoxim
proxetil.“ Clin. Pharm. Ther. 46 (1989) 647-685. |
| 16.
Guggenbichler J.P.: „Unpublizierte Beobachtungen bei
6 Säuglingen <3 Monate.“ 1996. |
| 17.
Fraschini F., Scaglione F., Demartini G.: Clarithromycin
clinical pharmacokinetics.“ Clin. Pharmacokinetics
25 (1993) 189-204. |
| 18.
Guggenbichler J.P., Tacacs F.: „Relative klinische
Bioverfügbarkeit der Kombination Trimethoprim-Sulfametrol
nüchtern und nach einer Mahlzeit bei Kindern.“
Päd. Pädol. 15 (1980) 305-320. |
| 19.
McCracken G.: „Pharmacologic evaluation of orally
administered antibiotics in infants and children: Effect
of feeding on bioavailability.“ Pediatrics 62 (1978)
738-743. |
| 20.
Petrick R.J., Kleimann K.: „Meal interference with
antibiotics administered orally in hospitals.“ Am.
J. Hosp. Pharm. 32 (1975) 1008-1013. |
| 21.
Elliott J.P.: „Effect of acute gastrointestinal disorders
on the bioavailability of antibiotics.“ Chemoth. Rev.
3 (1976) 254-268. |
| 22.
Parsons R.L., Paddock G.M.: „Drug absorption of two
antibacterial drugs cepha-lexin and co-trimoxazol in malabsorption
syndromes.“ J. Antimic. Chemoth. 1 Suppl (1975) 59-67. |
| 23.
Guggenbichler J.P.: „Resorption oral verabreichter
Antibiotika und Chemotherapeutika bei Kindern und ihre Beeinflussung.“
Päd. Praxis. 27 (1982) 669-695. |
| 24.
Kastner U., Guggenbichler J.P.: „Influence of Macrolide
Antibiotics on promotion of Resistance in the oral flora
of children.“ Infection 29 (2001) 251-256. |
| 25.
Black M.E., Hruby D.E.: „A single amino acid substitution
abolishes feedback inhibition of vaccinia virus thymidine
kinase.“ J. Biol. Chem. 267 (1992) 9743-9748. |
| 26.
Kearns G.L., Young R.A., Jacobs R.F.: „Cefotaxime
dosing in infants and children. Pharmacokinetic and clinical
rationale for an extended dosing interval.“ Clin.
Pharmacokinetics. 4 (1992) 284-297. |
| 27.
Raucy J.L., Allen S.W.: „Recent advances in P 450
Research.“ Pharmacogenomics J. 1 (2001) 178-186. |
| 28.
Periti P., Mazzei Th., Mini E., Novelli A.: „Pharmacokinetic
drug interactions of macrolides.“ Clin. Pharmacokinetics
23 (1992) 105-131. |
| 29.
Principi N., Esposito S.: „Comparative tolerability
of erythromycin and newer macrolide antibacterials in pediatric
patients.“ Drug Saf. 20 (1999) 25-41. |
| 30.
Autret E.: „Comparative safety and efficacy of cefpodoxime
proxetil and amoxicillin/clavulanic acid in the treatment
of acute maxillary sinusitis in children.“ 18th Intern.
Congress of Chemoth. 26 Suppl. E (1990) 29-34. |
| 31.
Chalmers J.P., Tiller D.J.: „Effects of treatment
on the mortality rate in septicemia.“ BMJ 2 (1969)
338-341. |
| 32.
Craven D.E., Kollisch N.R., Hsieh C.R.: „Vancomycin
treatment of bacteremia caused by oxacillin resistant Staphylococcus
aureus. Comparison between ß lactam antibiotic treatment
of bacteremia caused by oxacillin sensitive Staphylococcus
aureus.“ J. Infect. Dis. 147 (2001) 137-143. |
| 33.
Craig W.A.: „Choosing an antibiotic on the basis of
pharmacodynamics.“ Ear Nose Throat J. 77 Suppl. (1998)
7-11. |
| 34.
Craig W.A., Ebert S.C.: „Killing and re-growth of
bacteria in vitro. A review.“ Scand. J. Infect. Dis.
74 Suppl. (1991) 63-70. |
| 35.
Hanberger H., Nilsson L.E., Maller R., Nilsson M.: „Pharmacodynamica
of ß lactam antibiotics on Gram-negative bacteria:
initial killing, morphology and postantibiotic effect.“
Scan. J. Inf. Dis. Suppl. 74 (1991) 118 -123, |
| 36.
Begg E.J., Peddie B.A., Chambers S.T., Boswell D.R.: „Comparison
of gentamicin dosing regimens using an in vitro model.“
J. Antimic. Chemoth. 29 (1992) 427-433. |
| 37.
Guggenbichler J.P., Semenitz E., König P.: „Kill
kinetics and regrowth pattern of bacteria exposed to antibiotic
concentrations simulating those observed in vivo.“
J. Antimic. Chemoth. 15 Suppl. A (1985) 139-146. |
| 38.
Guggenbichler J.P., Allerberger F., Bonatti H., Semenitz
E., Dierich M.P.: „Absterbekinetik unter fluktuierenden
Konzentrationen von Antibiotika.“ Wiener Klin. Wochenschr.
101 (1989) 224-229. |
| 39.
König P., Guggenbichler J.P., Semenitz E., Foisner
W.: „Kill kinetics of bacteria under fluctuating concentrations
of various antibiotics. II description of experiments.“
Chemotherapy 32 (1986) 44-58. |
| 40.
Guggenbichler J.P.: „Eigene unpublizierte Beobachtungen“
(1991). |
| 41.
Vogelman B., Gudmundsson S., Leggett J.: „Correlation
of antimicrobial pharmacokinetic parameters with therapeutic
efficacy in an animal model.“ J. Inf. Dis. 158 (1988)
831-847. |
| 42.
Gengo F.M., Mannion T.W., Nightingale C.H., Schentag J.J.:
„Integration of pharmacokinetics and pharmacodynamics
of methicillin in curative treatment of experimental endocarditis.“
J. Antimicrob. Chemoth. 14 (1984) 619-631. |
| 43.
Dagan R., Leibovitz E., Greenberg D., Yagupsky P., Fliss
D.M., Leiberman A.: „Early eradication of pathogens
from middle ear fluid during antibiotic treatment of acute
otitis media is associated with improved outcome.“
Pediatric Infectious Dis. Journal 17 (1998) 778-782. |
| 44.
Gillespie M.B.: „Recurrent otitis media in children.“
JAMA 289 (2003) 1383-1384. |
| 45.
Gehanno P.: „Twice daily cefpodoxime proxetil compared
with thrice daily amoxicillin/clavulanic acid for treatment
of acute otitis media in children.“ Scan. J. Infect.
Dis. 26 (1994) 577-584. |
| 46.
Safran C.: „Cefpodoxime proxetil: dosage, efficacy
and tolerance in adults suffering from respiratory tract
infections.“ J. Antimic. Chemoth. Suppl. E (1990)
93-101. |
| 47.
Guggenbichler J.P.: „Eigene unpublizierte Beobachtungen“
(1991). |
Anschrift
des Verfassers:
Univ.-Prof. Dr. J. Peter Guggenbichler
Klinik mit Poliklinik der Universität Erlangen-Nürnberg
D-91054 Erlangen, Loschgestraße 15
E-Mail: prof.guggenbichler@gmx.de
|
|
|