| Kawasaki-Syndrom (Mukokutanes Lymphknotensyndrom) |
| W. Formanek Abteilung für Kinder- und Jugendheilkunde mit Infektionskrankheiten, Wilhelminenspital der Stadt Wien (Vorstand: Prim. Univ.-Prof. Dr. M. Götz) |
| Schlüsselwörter Zusammenfassung Key-words Summary Epidemiologie Klinik Kardiale Komplikationen Immunpathogenese des Kawasaki-Syndroms Superantigene Differentialdiagnosen Therapie Prognose Literatur |
| Schlüsselwörter:
Kawasaki-Syndrom, Vaskulitis, erworbene Herzerkrankung, intravenöses Immunglobulin, inkomplette oder atypische Fälle, Superantigenhypothese |
| Zusammenfassung Das Kawasaki-Syndrom (KS) wird zu der Krankheitsgruppe der primär systemischen Vaskulitiden gezählt, deren Ursache in der Gefäßwand selbst vermutet wird. Vor der Erstbeschreibung als eigenständiges Krankheitsbild 1967 durch Dr. Tomisaku Kawasaki wurde das KS als infantile Verlaufsform der Polyarteriitis nodosa in der Literatur erwähnt. Die akute febrile Vaskulitis befällt vor allem kleine Kinder und Säuglinge und ist heute in Industrieländern (noch vor dem rheumatischen Fieber) die häufigste Ursache erworbener Herzerkrankungen im Kindesalter. Die Krankheit beginnt mit einer Entzündung der Kapillaren, Arteriolen und Venolen der Haut. Nach wenigen Tagen werden die kleinen und mittleren Arterien mit Bevorzugung der mittleren und großen Koronararterien befallen. In der akuten Phase zeigt die histologische Untersuchung eine Perivaskulitis und Vaskulitis von Arteriolen, Kapillaren und Venolen, sowie eine Entzündung der Intima mittlerer und großer Arterien. Das entzündliche Infiltrat befällt zunächst das perivaskuläre Gewebe und greift dann auf Media und Intima über. In der subakuten Phase findet sich eine Panvaskulitis und Ödem der Gefäßwand mit Aneurysmenbildung, Thrombosen und Stenosen mittlerer Arterien. In der Rekonvaleszenzphase kann es zu Vernarbungen und Intimaverdickungen kommen. In Autopsiepräparaten fanden sich einerseits eine massiv mit Entzündungszellen infiltrierte Intima und Media mittlerer und großer Koronararterien, und anderer zentraler Gefäße, andererseits obstruierende Veränderungen durch Plättchenthromben. Thrombosen, Stenosen und Aneurysmen können sowohl im Bereich der Koronararterien, als auch im Bereich anderer Arterien, wie z.B. A. axillaris, -brachialis, -subclavia, A. iliaca, -femoralis, -poplitea, A. renalis oder der Zerebralgefäße auftreten. Die Einführung hochdosierter Immunglobulingaben in Kombination mit ASS in die Therapie des Kawasaki-Syndroms (KS) hat, innerhalb der ersten 10 Tage nach Fieberbeginn angewendet, geholfen, die Prävalenz von Koronararterienaneurysmen signifikant zu verringern. Rasches Erkennen und Behandeln sind dafür entscheidend; wobei sich die Diagnose mangels eines objektiven Labortests allein aufklinische Symptome stützt und letztlich vorläufig eine Ausschlußdiagnose bleibt. Die Häufigkeit inkompletter oder untypischer Fälle, die trotzdem mit einer Entwicklung von Koronararterienaneurysmen einhergehen, wird in verschiedenen Studien zwischen 10% und 45% angegeben. Viele atypische Fälle von Kawasaki-Syndrom (KS) sind erst durch die Entdeckung von Koronararterienaneurysmen mittels Echokardiographie oder postmortal, autoptisch diagnostiziert worden. Das größte Risiko ein atypisches KS zu entwickeln haben Säuglinge in den ersten 6 Lebensmonaten! Deshalb sollten auch Kinder mit persistierendem Fieber ungeklärter Ursache, einer akuten Phase-Reaktion und einem nur inkompletten klinischen Bild von Kawasaki-Syndrom (KS) im Zweifelsfalle intravenös mit Immunglobulin behandelt und einer Echokardiographie zugeführt werden. Obwohl die nach wie vor attraktive und plausible Superantigenhypothese viele Fragen zu beantworten vermag, ist die endgültige Beweisführung eines infektiösen Agens als Krankheitsursache des MCLS noch ausständig. |
| Key-words: Kawasaki syndrome, vaskulitis, acquired heart disease, intravenous immunoglobulin, incomplete or atypical cases, superantigen |
| Summary Kawaskai syndrome (KS) is an acute febrile multiorgan vasculitis predominantly affecting children younger than 5 years. It was originally described in Japan by Dr. Tomisaku Kawasaki in 1967. The illness is similiar to rare condition previously called infantile polyarteritis. The most important complication of Kawasaki syndrome (KS) is coronary arteritis. Kawasaki disease has surpassed rheumatic fever as the leading cause of acquired heart disease in children in developed countries. The introduction of high-dose intravenous immunoglobulin in combination with acetyl-salicylic acid treatment, within the first 10 days after the onset of fever, has significantly reduced the prevalence of coronary artery abnormalities in patients with Kawasaki syndrome. Early recognition and prompt treatment are therefore critical. Unfortunately, in the absence of a diagnostic test the current definition of Kawasaki syndrome (KS) is based on diagnostic criteria that use clinical signs and symptoms that overlap with other illnesses. The clinical criteria do not identify every case. Incomplete or atypical cases comprise between 10% and 45% of cases in published studies. Many incomplete cases have only come to light because coronary artery aneurysms have been found on Echocardiographie or at postmortem. Therfore presumptive treatment should be started in any child with a persistent fever, without other explanation, some of the clinical criteria of Kawasaki syndrome (KS) and an acute phase response even if the diagnosis is uncertain. Such patients should undergo imrnediate echocariography and then a repeat study 4-6 weeks after the onset of illness. The epidemiology of the disease strongly suggests an infectious etiology, but no single agent has been implicated to the disease. Whilst the superantigen hypothesis remains an attractive and plausible explanation, definitive evidence for the theory is still awaited. |
Epidemiologie Obwohl das KS weltweit bei Kindern aller Rassen diagnostiziert wird, kommt es am häufigsten in der japanischen Bevölkerung vor, mit einer Inzidenz von 5.000-6.000 Fällen pro Jahr in Japan (entspricht 76/100.000 < 5a alten Kindern in Japan). In den Vereinigten Staaten werden pro Jahr ca. 3.000 Patienten mit dieser Diagnose hospitalisiert. 80% aller Patienten sind jünger als vier Jahre. In den Vereinigten Staaten und Japan wurde von saisonalen (Spätwinter und Frühling, weniger im Spätherbst) und örtlichen Epidemien berichtet. InJapan wurde 1979, 1982 und 1985/86 eine epidemieartige Häufung beobachtet. In der Kinderinfektionsabteilung im Wilhelminenspital wurde im Jahr 1996 einmal, im Jahr 1997 und 1998 je zweimal die Diagnose MCLS gestellt. |
| Klinik Typisch für das KS ist ein dreiphasischer Verlauf, wobei
die akute Phase der Krankheit mit hohem Fieber bis 40°C und einem
häufig toxischen Erscheinungsbild (stark reduzierter AZ, Irritabilität, beeinträchtigte
Bewußtseinslage) beginnt und ohne antiinflammatorische Therapie 1-2 Wochen
anhält (Abb. 2).
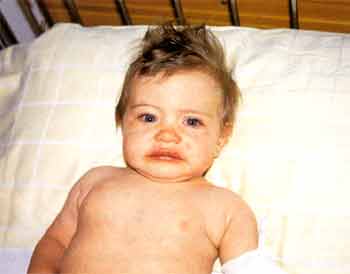
In 90 % der Patienten beginnt eine seröse Konjunktivitis kurz nach dem Fieberbeginn. Sie bezieht die Bulbusbindehaut mehr mit ein als die Augenlidbindehaut. Die Blutgefäße der Bindehaut erscheinen gestaut und verbreitert. Es finden sich keine eitrige Sekretion, kaum Schmerzen und kein Ödem. In 83% der Patienten ist die Konjunktivitis mit einer Iridozyklitis (Spaltlampe) vergesellschaftet (Abb. 1 ).
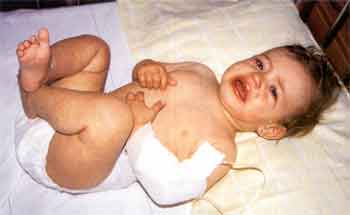
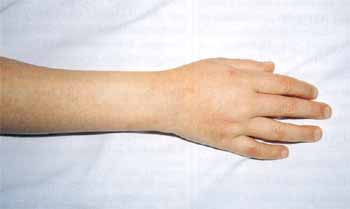
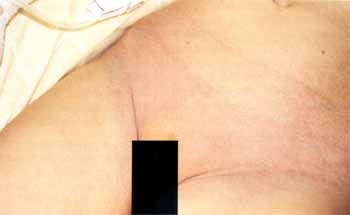
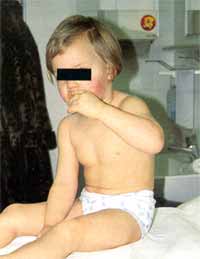
Eines der Frühsymptome des KS sind perineale Eruptionen (in einer Studie fand sich in 67% ein blasses, fleckiges Erythem im Bereich des Perineums) mit Beginn innerhalb der ersten sechs Tage (Abb. 5). Das in den meisten Fällen vorhandene Erythem der Hand- und Fußinnenflächen und Ödem der Hand- und Fußrücken, mit einer diffusen, nicht selten schmerzhaften Schwellung der Finger, beginnt gewöhnlich innerhalb einiger Tage nach Krankheitsbeginn (Abb. 4).
Mason und Takahashi haben die Häufigkeit der verschiedenen Symptome beim typischen wie auch atypischen KS untersucht (Tab. 1a), welches Ergebnis zu der Überlegung führen sollte, die Echokardiographie rechtzeitig in den diagnostischen Prozeß mit einzubeziehen; da das klinische Bild mit vielen anderen Krankheiten (am häufigsten Masern und Scharlach) verwechselt werden und außerdem oft nur bruchstückhaft vorhanden sein kann.
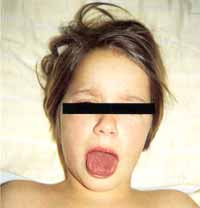
Rückfallsquote: Andere mit dem KS assoziierte klinische Bilder: In Ausnahmefallen wurde von dem Auftreten eines sensorineuralen Hörverlustes, zerebraler Krampfanfalle, Hirn- (V:a.N.facialis) oder peripherer Nervenlähmungen berichtet. Ein Hydrops der Gallenblase kann mit oder ohne kolikartigen Schmerzen ablaufen. Des weiteren können eine Hepatosplenomegalie sowie, relativ häufig Inappetenz, Übelkeit, Erbrechen, Durchfall und Bauchschmerzen auftreten. Als weitere Komplikationen wurden Aneurysmen großer peripherer Arterien oder durch Thrombosen verursachte Infarkte verschiedener Organe gesehen. |
| Kardiale
Komplikationen Möglich sind das Auftreten einer Peri-Myo-Endokarditis sowie einer koronaren Herzkrankheit mit den Folgen einer kardialen Insuffizienz oder Arrhythmien. Sterben kann der Patient in der akuten Phase an der Myokarditis, in der subakuten Phase an einem Herzinfarkt. Eine Myokarditis kann innerhalb der ersten Woche nach Fieberbeginn auftreten. Ein Perikarderguß wird gegen Ende der akuten Phase als Folge einer Peri- und/oder Myokarditis gefunden. Der Erguß führt selten zu einer Herzbeuteltamponade und bildet sich meist von selbst zurück. Eine Herzinsuffizienz wird in der akuten Phase durch eine Myokarditis und in der subakuten Phase, meist sekundär ischämisch bedingt, durch eine koronare Herzkrankheit verursacht. Veränderungen an den (vor allem großen) Koronararterien treten in
ca. 25% der Patienten auf, die nicht mit IVIG behandelt wurden. Aneurysmen der
Koronararterien werden mit Hilfe der Echokardiographie meist gegen Ende der ersten Woche
mit Gipfel um die dritte bis vierte Woche gefunden. Die akute Phase wird von der subakuten Phase gefolgt, die ungefähr 25 Tage lang anhält, während welcher Fieber, Exanthem und Lymphadenopathie sich zurückbilden, aber eine gesteigerte Irritabilität, Inappetenz und die seröse Konjunktivitis bestehen bleiben können. Während dieser Phase können eine periunguale Schuppung der Finger- und Zehenspitzen, Arthralgien und Arthritis, ein Gallenblasenhydrops wie auch palpable periphere Aneurysmen oder eine myokardiale Dysfunktion sich ausbilden. Die Rekonvaleszenz-Phase beginnt, wenn sich die klinischen Symptome zurückgebildet haben, und dauert an, bis sich die Blutsenkungsgeschwindigkeit normalisiert hat, üblicherweise bis 70 Tage nach Krankheitsbeginn. Von den Laborveränderungen (siehe Tabelle 2) läßt sich, einzeln betrachtet, bei der Diagnosestellung des KS, abgesehen von einer regelmäßig vorhandenen mäßigen Leukozytose, auch keine hundertprozentige Hilfe erwarten, wie die Tabelle 2a zeigt. Eher typisch ist eine Kombination der verschiedenen tabellarisch angeführten Befunde, sowie die Abwesenheit einer den schlechten klinischen Zustand erklärenden anderen Erkrankung.
EKG-Veränderungen: treten in über der Hälfte aller Patienten mit MCLS auf: verlängertes PR-Intervall, Q- Wellen, unspezifische ST-T-Wellen-Veränderungen, links-ventrikuläre Hypertrophie, ventrikuläre Arrhythmien. Eine Echokardiographie sollte bei allen Patienten mit MCLS vor dem 7. Krankheitstag und 6-8 Wochen später, bei kleineren Säuglingen und anderen Risikopatienten eventuell schon früher (nach 2-3 Wochen) durchgeführt werden. Wenn mit Hilfe der zweidimensionalen Echokardiographie kein Aneurysma darstellbar ist, so ist auch das Vorhandensein einer Stenose äußerst unwahrscheinlich und eine weitere Diagnostik in der Regel nicht indiziert. Kinder mit normalen Herzechos oder nur kleinen Aneurysmen, die sich innerhalb von sechs Monaten zurückbilden, benötigen keine invasiven Nachuntersuchungen. Kinder, deren Aneurysmen 6 Monate nach Diagnosestellung persistieren, sollten 6-12 Monate nach Erkrankungsbeginn angiographiert werden, um Stenosen aufzudecken. Solche mit Riesenaneurysmen, Klappendefekten oder Ischämiezeichen müssen früher angiographiert werden. In den Kontrollen dieser letzten Patienten kann auch eine Fahrradergometrie und mehr noch eine Dobutamin-Stress-Echocardiographie (vor allem bei sehr kleinen Kindern) sowie eine Thallium-Szintigraphie in der Aufdeckung von Ischämien hilfreich sein. Als Risikofaktoren für die Entwicklung von Koronararterienaneurysmen oder Thrombosen gelten: ein typisches, komplettes klinisches Bild, ein Erkrankungsrezidiv, das männliche Geschlecht, ein Alter unter einem Jahr und über sieben Jahren, eine Tachykardie (mittlere 24h-HF), ein Komplementverbrauch [CHSO, C3, C4-Abnahme und C3d ( -Komplementspaltprodukt) -Zunahme], eine Thrombozytopenie (Fibrinogenabnahme, Fibrinspaltproduktezunahme), ein hohes CRP und hohe Leukozytenzahlen (beides v.a. bei Kindern im ersten Lebensjahr), ein niedriger Serumkaliumspiegel, sowie eine späte oder zu niedrig dosierte Gabe von intravenösem Immunglobulin. |
| Immunpathogenese des Kawasaki-Syndroms Die Ursache der Erkrankung ist nach wie vor unbekannt. Für einen Infektionserreger als Auslöser des MCLS spricht: 1. akute, selbst limitierte Natur der Erkrankung |
| Superantigene Die Superantigenhypothese versucht die beim KS anzutreffenden immunologischen Veränderungen dadurch zu erklären, daß im Falle einer angenommenen Stimulation des Immunsystems durch Superantigene die physiologischerweise sehr spezifische (und deshalb auf wenige T-Zellen beschränkte) Antigenerkennung durch T-Zellen unspezifischer erfolgt, sodaß ein Superantigen von einer ungewöhnlich großen Zahl von T-Zellen als Antigen erkannt werden kann. Der Terminus Superantigen beschreibt eine Gruppe von Antigenen, die sich in verschiedener Hinsicht von typischen Protein- oder Peptid-Antigenen unterscheiden. Es sind zwei Arten von mikrobiellen Superantigenen bekannt: T-Zell- Superantigene und B-Zell-Superantigene. Eine mögliche Wirkung als T-Zell-Superantigen wird angenommen von: Staphylococcus aureus (für das KS am besten untersucht): pyrogene Toxine, Enterotoxine, exfoliatives Toxin A und B, Toxic Shock Syndrome Toxin 1; pyrogene Toxine, M-Protein, Streptokokken-Superantigen SSA, mitogener Faktor; Yersinia pseudo-tuberculosis, Yersinia enterocolitica; Clostridium perlringens; Pseudomonas aeruginosa; Mycobacterium tuberculosis; Toxoplasma gondii; Mycoplasma arthritidis; Streptokokken der Gruppe A, Tollwut-Virus; CMV; EBV. T-Zell-Superantigene vermögen eine große Zahl von T-Zellen (unspezifisch) zu stimulieren, indem sie:
(T-Zellen können Antigene mit Hilfe ihres T-Zell-Rezeptors nur dann als solche erkennen, wenn diese der T-Zelle gemeinsam mit körpereigenen MHC-Molekülen auf der Oberfläche der antigenpräsentierenden Zelle angeboten werden. )
Die Antigenerkennung durch T-Lymphozyten erfolgt über den T-Zell-Antigenrezeptor, welcher aus zwei unterschiedlichen polymorphen Glykoproteinen gebildet wird. Die vier bekannten Antigenrezeptorketten (alpha, beta, gamma, delta) bilden auf der T-Zell-Oberfläche alpha/beta oder gamma/delta-Heterodimeren. Eine einzelne T-Zelle exprimiert ca. 30.000 Antigenrezeptoren eines Typs (alpha/beta in der Mehrzahl der T-Zellen oder gamma/delta in 5-15% aller T-Zellen) und einer einzigen Spezifität. Der extrazelluläre Abschnitt der Antigenrezeptorketten besteht (wie bei den Immunglobulinen) aus einer variablen und einer konstanten Region, wobei die variable Region der beiden Ketten der spezifischen Antigenerkennung dient. Die variable Region der alpha-Kette wird durch je ein Gen aus zwei Gen-Familien (den 70 Variablen [V-] und 61 Joining [J-] Gensegmenten) auf Chromosom 14 kodiert, während für die variable Region der ß-Kette je ein Gen aus drei Gen-Familien (52 V-, 2 Diversity [(D-)], und 13 J-Gene) auf Chromosom 7 im humanen Genom zur Auswahl bereitsteht. Im Falle eines T-Zell-Superantigens sind für die Spezifität der Antigenerkennung durch den T-Zell-Rezeptor ausschließlich die V-Gene der variablen Region der ß-Kette, nicht aber die übrigen (D- und J-) Gene der variablen Region der ß-Kette oder die (V- und J- ) Gene der variablen Region der alpha-Kette verantwortlich. Deshalb wird ein T-Zell-Superantigen von praktisch allen T-Zellen als Antigen erkannt, die ein bestimmtes spezifisches Vß-Segment auf ihrem T-Zell-Rezeptor tragen, unabhängig davon, wie die restliche ß-Kette oder die alpha-Kette ihres T-Zell-Rezeptors zu dem Antigen paßt. Im Gegensatz zu Superantigenen benötigen normale Peptid-Antigene alle 5 variablen Elemente der alpha- und ß-Kette des T-Zell-Rezeptors zur optimalen T-Zell-Erkennung und stimulieren dadurch nur eine begrenzte Zahl von T-Zellen. Beim KS kommt es vor allem zu einer Zunahme Vß2-positiver T-Zellen (wie auch beim Toxic Shock Syndrome), welcher Befund gut zu der These einer Superantigen-Stimulation des Immunsystems bei diesen beiden Krankheiten passen würde. In obduzierten Patienten mit MCLS haben sich ebenso im Myokard wie auch in Koronararterienaneurysmen eine selektive Expansion von TCRßV2+T-Zellen sowohl in CD4+ als auch in CD8+ -T-Zell-Myokard-Infiltraten finden lassen. Leider haben andere Studien solche Veränderungen nicht finden können. Es ist unklar, ob diese widersprüchlichen Ergebnisse auf methodologischen Unterschieden, auf unterschiedlichen Krankheitszeitpunkten, zu denen die Untersuchungen durchgeführt wurden, oder auf ein konventionelles Antigen statt eines Superantigens als Krankheitsauslöser beruhen. Die uneingeschränkte T-Zell-Aktivierung führt zur gesteigerten Freisetzung von Zytokinen, wie Tumornekrosefaktor-alpha, und vielen anderen mit den Folgesymptomen einer ausufernden, sich selbst unterhaltenden Entzündungsreaktion (Fieber bis kardiogener Schock). Es scheint, daß das KS nicht von einem einzigen, sondern von verschiedenen bakteriellen, viralen oder parasitären Toxinen getriggert werden kann, vielleicht auch von einem ubiquitären infektiösen Agens, dem der Mensch im Laufe seines Lebens regeImäßig ausgesetzt ist und wogegen er sich früh in der Kindheit eine Immunität erwirbt. Möglicherweise befällt dieses Krankheitsbild vor allem deshalb Kinder zwischen einem halben und vier Jahren, weil es dieser Altersgruppe an adäquaten protektiven Ak. mangelt. Bei der Krankheitsentstehung scheint aber auch eine genetische Disposition eine Rolle zu spielen, wie die größere Häufigkeit des MCLS bei Kindern japanischer Eltern unabhängig von ihrem Wohnort sowie die Häufung der HLA-Antigene Bw22, B22, B22J2 in Japan und Bw51 bei europäischen Patienten mit MCLS vermuten läßt. Immunologisch findet sich eine ausgeprägte Aktivierung von B- und T-Zellen mit einer deutlichen Vermehrung von CD4-positiven T-Zellen und einer Erhöhung der CD4/CD8-Ratio. Des weiteren kommt es während der akuten Krankheitsphase zu einer Zunahme der Serum-Zytokinkonzentrationen, insbesondere von IL-1, lösl. IL2-Rezeptor, IL-6, TNF-alpha und IFN-gamma, wie auch von CD23. Als weiteres zusätzliches Charakteristikum findet sich während der ersten 10 Tage der akuten Phase eine Vermehrung des Serum-IgE, von IgM und Akute-Phase-Proteinen. Als Besonderheit konnten IgM nachgewiesen werden, die mit Interferon-gamma, IL-I beta oder TNF-alpha vorbehandelte Endothelzellen zu lysieren vermögen. Die pathogenetische Rolle ebenfalls gefundener zirkulierender Immunkomplexe bleibt vorerst unklar. Bei schwerem Verlauf kommt es zu einem Verbrauch von Komplement mit Erniedrigung von C3 und C4 und Erhöhung des Spaltproduktes C3d. Die initiale Gefäßläsion im KS geht mit einer Aktivierung der Gefäßendothelzelle einher, die zur Expression von Leukozyten-Adhäsionsmolekülen führt, welche die Bildung von Zytokinen induzieren und zur Infiltration von aktivierten CD4+ und CD8+ T-Zellen genauso wie Monozyten/Makrophagen führen. Im peripheren Blut steigt die Zahl aktivierter T- und B-Zellen, Monozyten und Makrophagen. Die Wirkung der IVIG beim KS dürfte über darin enthaltene spezifische neutralisierende Antikörper gegen Superantigen-Toxine und damit bewirkte Reduzierung der zytokininduzierten Endothelzellaktivierung zu suchen sein. |
| Differentialdiagnosen Urethritis, Enteritis oder meningeale Reizung können lokalisierte bakterielle oder virale Infekte vortäuschen. Virusinfekte wie EBV-Infektion, Masern, Adeno- oder Influenzavirusinfekte, Scharlach, akutes rheumatisches Fieber, Staphylokokken oder Streptokokken verursachtes Toxic Shock Syndrome, Leptospirose, JRA, Rocky Mountain spotted fever, Erythema multiforme, Drug reaction, Stevens-Johnson-Syndrome. Am ehesten macht die Abgrenzung der viszeralen Form der juvenilen rheumatoiden Arthritis, des Still-Syndroms, Probleme. Bei dieser Erkrankung tritt jedoch keine konjunktivale Injektion und keine so ausgeprägte zervikale Lymphadenopathie auf, das Exanthem ist flüchtig und diskret, Palmar und Plantarerythem so wie Erdbeerzunge und Lacklippen fehlen in der Regel, und Koronaraneurysmen oder Herzinfarkte werden nicht beobachtet. |
| Therapie wird in Tabelle 3 beschrieben. Obwohl die parenterale Gammaglobulintherapie generell als sicher gilt, wurden als gelegentliche Nebenwirkungen beschrieben: Anaphylaxie, Schüttelfrost, Fieber, Kopf- und Muskelschmerzen, Immunhämolyse, aseptische Meningitis, Serumkrankheit, Hepatitis C-Virusinfektion. Das Ansprechen auf die Gammaglobulintherapie ist meist dramatisch. Ist der Patient 48h nach der Verabreichung fieberfrei, bzw. im Allgemeinzustand wesentlich gebessert, wird die Gabe in derselben Dosis wiederholt.
|
| Prognose Die Letalität des MCLS wurde ursprünglich mit 1-2% angegeben, beträgt zur Zeit in Japan aber nur noch 0,4%. Die glücklicherweise seltenen Riesenkoronararterienaneurysmen mit einem DM von > als 9 mm (1% aller Patienten) haben nach wie vor eine ernste Prognose. Von diesen Patienten entwickeln fast die Hälfte eine Stenose oder komplette Obstruktion. 2/3 der Patienten mit Stenose entwickeln einen Herzinfarkt. Im Gegensatz zu Riesenaneurysmen bilden sich kleine oder mittlere Aneurysmen großteils wieder vollständig zurück, wobei die Sorge bestehen bleibt, ob diese Koronararterien nicht auch nach der Aneurysma-Rückbildung abnormal bleiben. Suzuki et al. untersuchten 23 Patienten in einem Follow-up mit
wiederholten Angiographien und intravaskulärem Ultraschall, um die Dicke der Intima und
Media der Arterienwand zu messen. Ebenso war die Reaktivität der Koronararterien auf Nitroglyzerin oder Dipyridamol im Bereich bestehender wie ehemaliger Aneurysmen reduziert. Solche Ergebnisse führen zu Überlegungen, ob diese verdickten und wenig reagiblen Arterien nicht gefährdet sein könnten, später eine Arteriosklerose mit allen Spätfolgen zu entwickeln. Welche Frage zu klären einer zukünftigen verstärkten Zusammenarbeit zwischen Pädiatern und Internisten überlassen bleibt. |
| Literatur: 1. Nelson: " Textbook of Pediatrics." 15th Edition, W.B. Saunders Company, 1996. 2. Wahn U., Wahn V, Seger R.: "Pädiatrische Allergologie und Immunologie." 3. Auflage, Urban & Fischer. 3. Stiehm E.R.: "Immunologic Disorders in Infants & Children." 4th Edition, WB. Saunders Company. 4. Bradley J., McCluskey J.: "Clinical Immunology." Oxford University Press, 1997. 5. Love P.E., Shores E.W.: "Antigen Receptor Signalling in T Cell Development and Selection." Seminars in Immunology, Academic Press, Volume 11, Issue4 (Aug 1999) 11-4. 6. Pamer E.G.: "Antigen Presentation in the Immune Response to Infectious Diseases" Clinical Infectious Diseases 28 (1999) 714-6. 7. Leung D.Y.M., Schliefertand P.M., Meissner H.C.: "The Immunpathogenesis and Management of Kawasaki Syndrome." Arthritis & Rheumatism, Vol. 4, No.9, Sept. (1998) 1538-1547. 8. Curtis N., Levin M.: Kawasaki disease thirty years on." Current Opinion in Pediatrics 10 (1998) 24-33. 9. Mason WH., Takahashi M.: "Kawasaki Syndrome." Clinical Infectious Diseases 28 (1999) 169-87. 10. Koyanagi H., Nakamura Y., Yanagawa H.: "Lower level of serum potassium and higher level of C-reactive protein as an independent risk factor for giant aneurysms in Kawasaki disease." Acta. Paediatr. 87 (1998) 32-6. 11. Suzuki Y., liJima M., Sasaki H., Muto T., Tanaka H., Kaneko K., Yamashiro Y.: "Tachycardia as a potential risk indicator for coronary arteriallesions in Kawasaki disease." Eur. J. Pediatr. 158 (1999) 207-209. 12. Zhang T., Yanagawa H., Oki I., Nakamura Y., Yashiro M., Ojima T., Tanihara S.: "Factors related to cardiac sequelae of Kawasaki disease." Eur. J. Pediatr. 158 (1999) 694-697. 13. Schiller B., Elinder G.: "Inflammatory parameters andsoluble cell adhesion molecules in Swedish children with Kawasaki disease : relationship to cardiac lesions and intravenous immunoglobulin treatment." Acta. Pediatr. 88 (1999) 844-8. 14. Bums J.C., Capparelli E. V, Brown J.A., Newburger J.W., Glode M.P.: "Intravenous gammaglobulin treatment and retreatment in Kawasaki disease." Pediatr. Infect. Dis. J. 17, Vol. 17, No. 12 (1998) 1144-8. 15. Newburger J.W.: "Treatment of Kawasaki disease." Lancet 347 (1996) 1128. 16. Kato H., Sugimura T., Akagi T.: "Long-term consequences of Kawasaki disease. A 10- to 21- year follow-up study of 594 patients." Circulation 94(1996) 1379-1385. 17. Wann E.R., Fehringer A.P., Ezepchuk Y. V, Schlievert P.M., Bina P., Reiser R.F., Höök M.M., Leung D.Y.M.: "Staphylococcus aureus Isolates from Patients with Kawasaki Disease Express High Levels of Protein A." Infection and Immunity, Vol. 67, No.9 (1999) 4737-4743. 18. Juvonen T., Juvonen J., Savolainen M.J.: "Is vasculitis a significant component of athero-sclerosis? Current Opinion in Rheumatology 11 (1999) 3-10. |
| Anschrift des Verfassers: Dr. W. Formanek Abteilung für Kinder- und Jugendheilkunde mit Infektionskrankheiten, Wilhelminenspital A-1171 Wien, Montleartstraße 37 |